
Bioinformatics and Experimental Analyses Reveal NFIC as an Upstream Transcriptional Regulator for Ischemic Cardiomyopathy

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
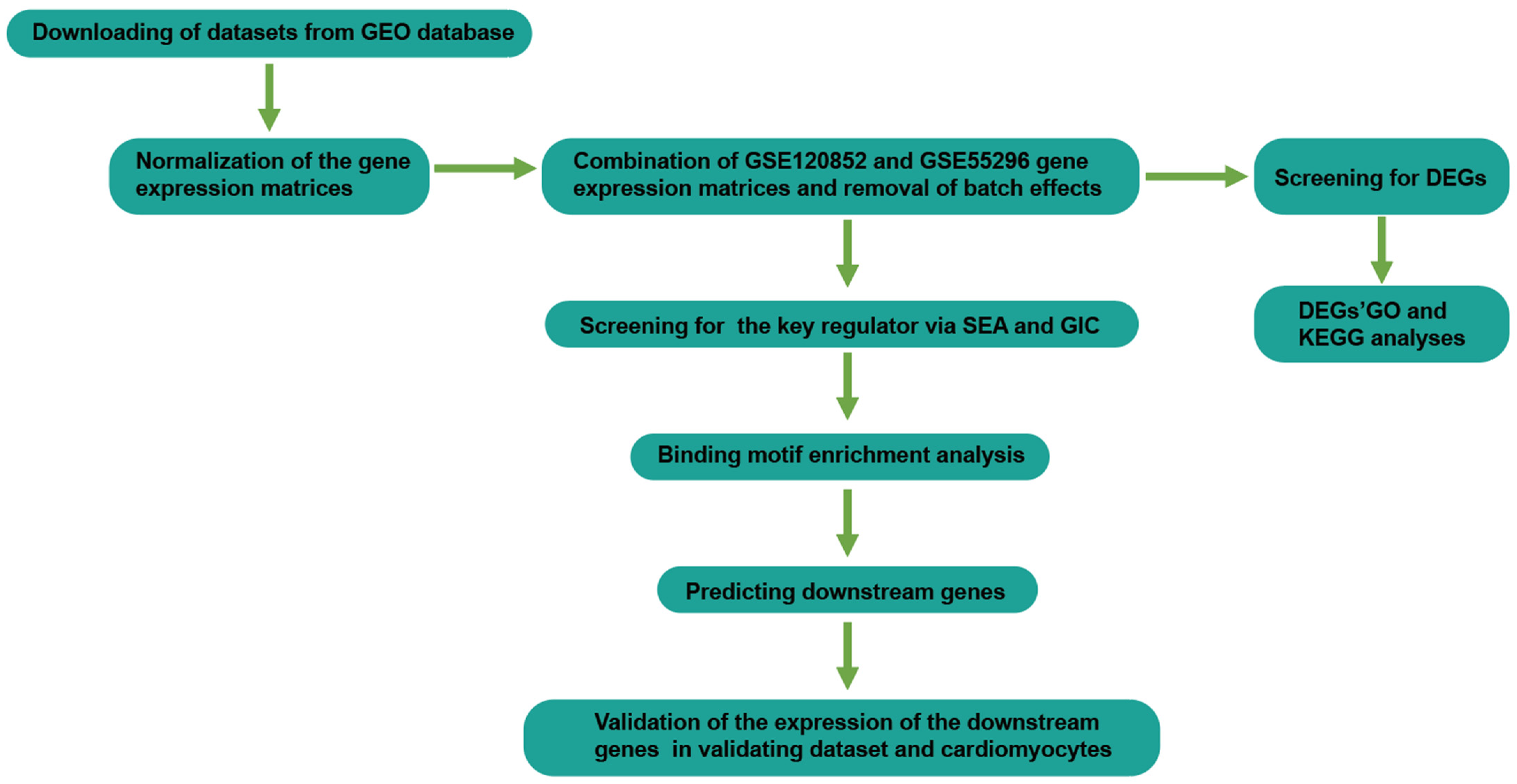
2. Materials and Methods
2.1. Data Selection
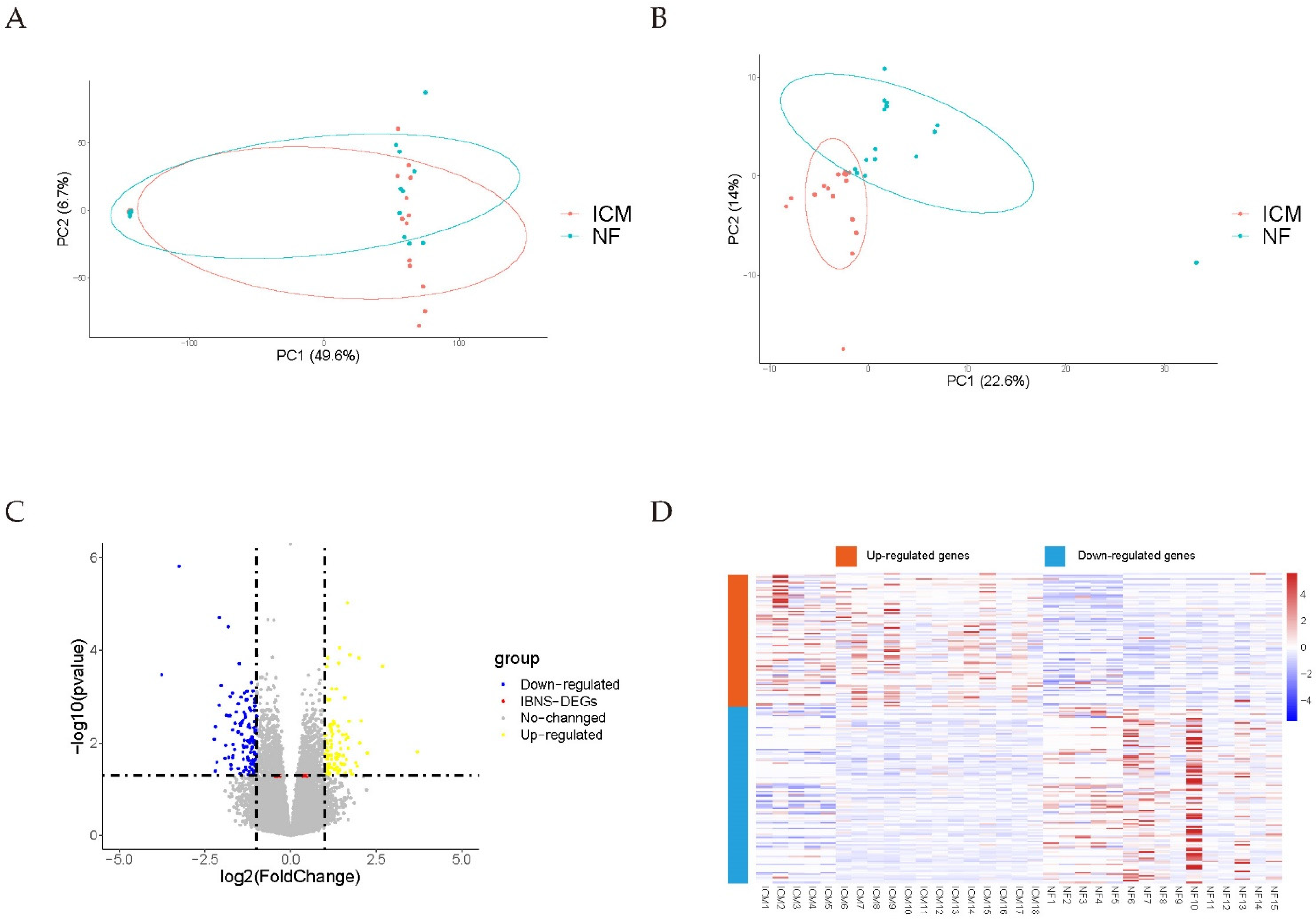
2.2. Data Preprocessing and DEGs Screening
2.3. GO and KEGG Functional Enrichment Analyses
2.4. Transcription Factor Binding Site Analysis
2.5. Construction of Lentiviral Vectors and Cell Culture
2.6. Quantitative Real-Time PCR (qRT-PCR)
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
3.1. Data Preprocessing and DEGs Screening
3.2. GO and KEGG Analyses of Downregulated Genes
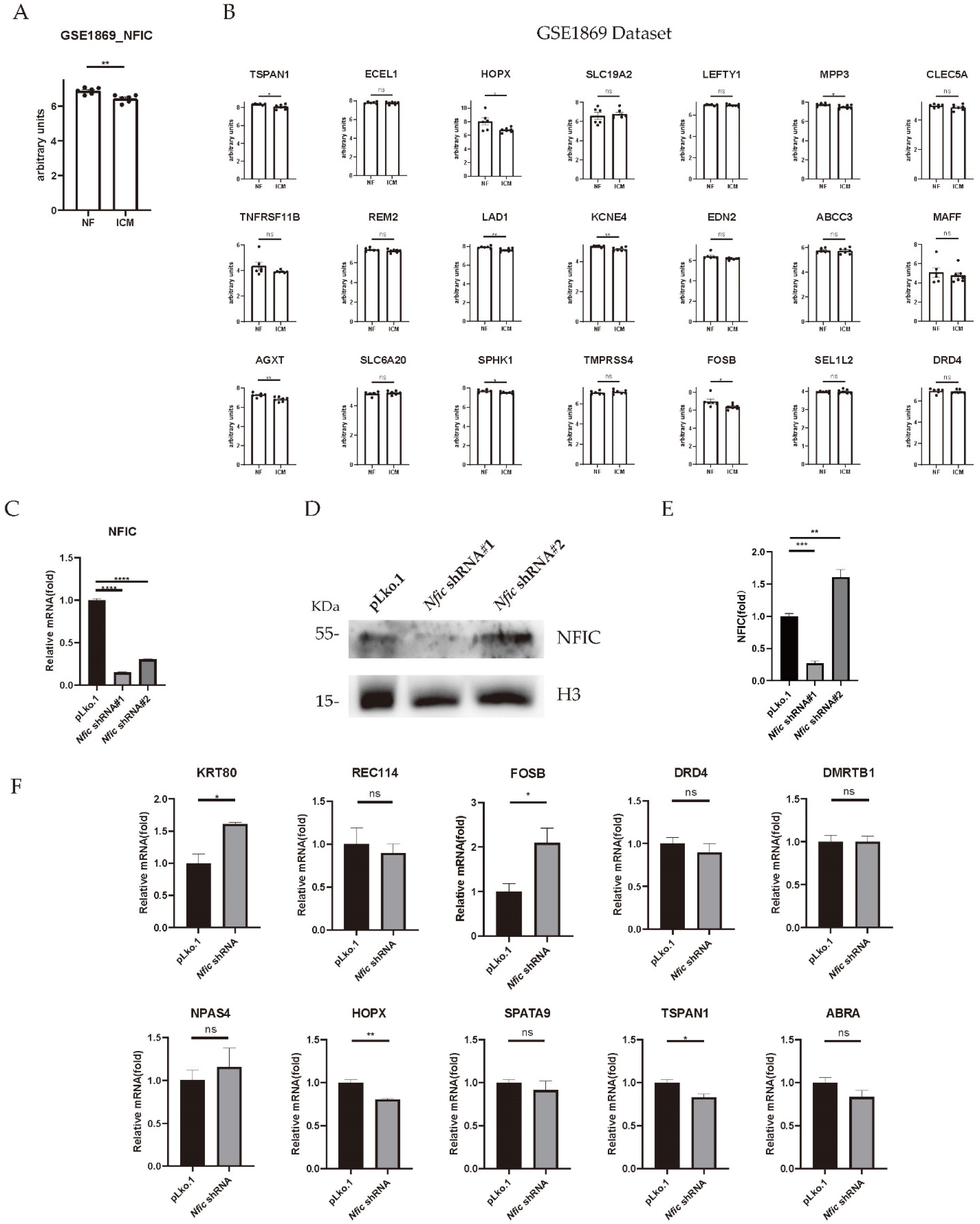
3.3. Enrichment Analysis of NFIC-Binding Motif on Downregulated Promoter Sequences
3.4. Verification of Predicted Genes with NFIC-Binding Motif by Validating Microarray Dataset and Nfic Knockdown Cardiomyocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Moroni, F.; Gertz, Z.; Azzalini, L. Relief of Ischemia in Ischemic Cardiomyopathy. Curr. Cardiol. Rep. 2021, 23, 80. [Google Scholar] [CrossRef]
- Sun, R.; Li, X.; Liu, M.; Zeng, Y.; Chen, S.; Zhang, P. Advances in stem cell therapy for cardiovascular disease (Review). Int. J. Mol. Med. 2016, 38, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Bian, J.; Popovic, Z.B.; Benejam, C.; Kiedrowski, M.; Rodriguez, L.L.; Penn, M.S. Effect of cell-based intercellular delivery of transcription factor GATA4 on ischemic cardiomyopathy. Circ. Res. 2007, 100, 1626–1633. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Liu, J.; Jiao, J.; Long, B.; Li, Q.; Tan, W.; Li, P. Transcription factor Foxo3a prevents apoptosis by regulating calcium through the apoptosis repressor with caspase recruitment domain. J. Biol. Chem. 2013, 288, 8491–8504. [Google Scholar] [CrossRef] [Green Version]
- Nagata, K.; Guggenheimer, R.A.; Enomoto, T.; Lichy, J.H.; Hurwitz, J. Adenovirus DNA replication in vitro: Identification of a host factor that stimulates synthesis of the preterminal protein-dCMP complex. Proc. Natl. Acad. Sci. USA 1982, 79, 6438–6442. [Google Scholar] [CrossRef] [Green Version]
- Mason, S.; Piper, M.; Gronostajski, R.M.; Richards, L.J. Nuclear factor one transcription factors in CNS development. Mol. Neurobiol. 2009, 39, 10–23. [Google Scholar] [CrossRef]
- Gronostajski, R.M.; Adhya, S.; Nagata, K.; Guggenheimer, R.A.; Hurwitz, J. Site-specific DNA binding of nuclear factor I: Analyses of cellular binding sites. Mol. Cell. Biol. 1985, 5, 964–971. [Google Scholar] [CrossRef]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-binding specificities of human transcription factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Shu, T.; Butz, K.G.; Plachez, C.; Gronostajski, R.M.; Richards, L.J. Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice. J. Neurosci. 2003, 23, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Steele-Perkins, G.; Plachez, C.; Butz, K.G.; Yang, G.; Bachurski, C.J.; Kinsman, S.L.; Litwack, E.D.; Richards, L.J.; Gronostajski, R.M. The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol. Cell. Biol. 2005, 25, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Driller, K.; Pagenstecher, A.; Uhl, M.; Omran, H.; Berlis, A.; Grunder, A.; Sippel, A.E. Nuclear factor I X deficiency causes brain malformation and severe skeletal defects. Mol. Cell. Biol. 2007, 27, 3855–3867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.C.; Herr, Y.; Kim, H.J.; Gronostajski, R.M.; Cho, M.I. Nfic gene disruption inhibits differentiation of odontoblasts responsible for root formation and results in formation of short and abnormal roots in mice. J. Periodontol. 2007, 78, 1795–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.S.; Park, J.T.; Kim, H.M.; Ko, J.S.; Son, H.H.; Gronostajski, R.M.; Cho, M.I.; Choung, P.H.; Park, J.C. Nuclear factor I-C is essential for odontogenic cell proliferation and odontoblast differentiation during tooth root development. J. Biol. Chem. 2009, 284, 17293–17303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.J.; Lee, H.K.; Park, S.J.; Cho, Y.S.; Bae, H.S.; Cho, M.I.; Park, J.C. Zinc balance is critical for NFI-C mediated regulation of odontoblast differentiation. J. Cell Biochem. 2012, 113, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Rau, A.; Marot, G.; Jaffrezic, F. Differential meta-analysis of RNA-seq data from multiple studies. BMC Bioinform. 2014, 15, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, E.; Hernandez, A.V.; Kattan, M.W. Meta-analysis: Its strengths and limitations. Cleve Clin. J. Med. 2008, 75, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; He, J.; Zhang, Z.; Jiang, S.; Gao, Y.; Pan, Y.; Wang, H.; Zhuang, L. Integrated Bioinformatics Analysis Reveals Marker Genes and Potential Therapeutic Targets for Pulmonary Arterial Hypertension. Genes 2021, 12, 1339. [Google Scholar] [CrossRef] [PubMed]
- Alimadadi, A.; Aryal, S.; Manandhar, I.; Joe, B.; Cheng, X. Identification of Upstream Transcriptional Regulators of Ischemic Cardiomyopathy Using Cardiac RNA-Seq Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 3472. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.F.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, X.L.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, R.; Cui, Q. Toward comprehensive functional analysis of gene lists weighted by gene essentiality scores. Bioinformatics 2021, 37, 4399–4404. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Berhanu Lemma, R.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Manosalva Perez, N.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022, 50, D165–D173. [Google Scholar] [CrossRef]
- Tzimas, C.; Rau, C.D.; Buergisser, P.E.; Jean-Louis, G., Jr.; Lee, K.; Chukwuneke, J.; Dun, W.; Wang, Y.; Tsai, E.J. WIPI1 is a conserved mediator of right ventricular failure. JCI Insight 2019, 5, e122929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarazon, E.; Rosello-Lleti, E.; Rivera, M.; Ortega, A.; Molina-Navarro, M.M.; Trivino, J.C.; Lago, F.; Gonzalez-Juanatey, J.R.; Orosa, P.; Montero, J.A.; et al. RNA Sequencing Analysis and Atrial Natriuretic Peptide Production in Patients with Dilated and Ischemic Cardiomyopathy. PLoS ONE 2014, 9, e90157. [Google Scholar] [CrossRef]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Chen, J.; Meng, Y.H.; Zhou, Y.; Yang, J.C.; Cui, Q.H. Defining Essentiality Score of Protein-Coding Genes and Long Noncoding RNAs. Front. Genet. 2018, 9, 380. [Google Scholar] [CrossRef] [PubMed]
- Morano, M.; Angotti, C.; Tullio, F.; Gambarotta, G.; Penna, C.; Pagliaro, P.; Geuna, S. Myocardial ischemia/reperfusion upregulates the transcription of the Neuregulin1 receptor ErbB3, but only postconditioning preserves protein translation: Role in oxidative stress. Int. J. Cardiol. 2017, 233, 73–79. [Google Scholar] [CrossRef]
- Pilz, P.M.; Hamza, O.; Gidlof, O.; Goncalves, I.F.; Tretter, E.V.; Trojanek, S.; Abraham, D.; Heber, S.; Haller, P.M.; Podesser, B.K.; et al. Remote ischemic perconditioning attenuates adverse cardiac remodeling and preserves left ventricular function in a rat model of reperfused myocardial infarction. Int. J. Cardiol. 2019, 285, 72–79. [Google Scholar] [CrossRef]
- Rotter, D.; Grinsfelder, D.B.; Parra, V.; Pedrozo, Z.; Singh, S.; Sachan, N.; Rothermel, B.A. Calcineurin and its regulator, RCAN1, confer time-of-day changes in susceptibility of the heart to ischemia/reperfusion. J. Mol. Cell. Cardiol. 2014, 74, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zhou, Y.; Gu, X.; Zhang, X.; Jia, Z. NLRX1/FUNDC1/NIPSNAP1-2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 2021, 54, e12986. [Google Scholar] [CrossRef]
- Zhang, H.; Xiao, Y.; Nederlof, R.; Bakker, D.; Zhang, P.; Girardin, S.E.; Hollmann, M.W.; Weber, N.C.; Houten, S.M.; van Weeghel, M.; et al. NLRX1 Deletion Increases Ischemia-Reperfusion Damage and Activates Glucose Metabolism in Mouse Heart. Front. Immunol. 2020, 11, 591815. [Google Scholar] [CrossRef]
- Alyahya, A.M.; Al-Masri, A.; Hersi, A.; El Eter, E.; Husain, S.; Lateef, R.; Mawlana, O.H. The Effects of Progranulin in a Rat Model of Acute Myocardial Ischemia/Reperfusion are Mediated by Activation of the P13K/Akt Signaling Pathway. Med. Sci. Monit. Basic Res. 2019, 25, 229–237. [Google Scholar] [CrossRef]
- Muszbek, L.; Yee, V.C.; Hevessy, Z. Blood coagulation factor XIII: Structure and function. Thromb. Res. 1999, 94, 271–305. [Google Scholar] [CrossRef]
- Ansani, L.; Marchesini, J.; Pestelli, G.; Luisi, G.A.; Scillitani, G.; Longo, G.; Milani, D.; Serino, M.L.; Tisato, V.; Gemmati, D. F13A1 Gene Variant (V34L) and Residual Circulating FXIIIA Levels Predict Short- and Long-Term Mortality in Acute Myocardial Infarction after Coronary Angioplasty. Int. J. Mol. Sci. 2018, 19, 2766. [Google Scholar] [CrossRef] [Green Version]
- Gemmati, D.; Vigliano, M.; Burini, F.; Mari, R.; El Mohsein, H.H.; Parmeggiani, F.; Serino, M.L. Coagulation Factor XIIIA (F13A1): Novel Perspectives in Treatment and Pharmacogenetics. Curr. Pharm. Des. 2016, 22, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Dardik, R.; Solomon, A.; Loscalzo, J.; Eskaraev, R.; Bialik, A.; Goldberg, I.; Schiby, G.; Inbal, A. Novel proangiogenic effect of factor XIII associated with suppression of thrombospondin 1 expression. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1472–1477. [Google Scholar] [CrossRef] [Green Version]
- Nahrendorf, M.; Aikawa, E.; Figueiredo, J.L.; Stangenberg, L.; van den Borne, S.W.; Blankesteijn, W.M.; Sosnovik, D.E.; Jaffer, F.A.; Tung, C.H.; Weissleder, R. Transglutaminase activity in acute infarcts predicts healing outcome and left ventricular remodelling: Implications for FXIII therapy and antithrombin use in myocardial infarction. Eur. Heart J. 2008, 29, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Louis Sam Titus, A.S.C.; Yusuff, T.; Cassar, M.; Thomas, E.; Kretzschmar, D.; D’Mello, S.R. Reduced Expression of Foxp1 as a Contributing Factor in Huntington’s Disease. J. Neurosci. 2017, 37, 6575–6587. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Liu, X.; Chai, N.; Lv, L.; Wang, R.; Li, X.; Nie, Y.; Shi, Y.; Fan, D. The transcription factor FOXO4 is down-regulated and inhibits tumor proliferation and metastasis in gastric cancer. BMC Cancer 2014, 14, 378. [Google Scholar] [CrossRef] [Green Version]
- Steele-Perkins, G.; Butz, K.G.; Lyons, G.E.; Zeichner-David, M.; Kim, H.J.; Cho, M.I.; Gronostajski, R.M. Essential role for NFI-C/CTF transcription-replication factor in tooth root development. Mol. Cell. Biol. 2003, 23, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Van Bortle, K.; Zhang, Y.; Zhao, M.T.; Zhang, J.Z.; Geller, B.S.; Gruber, J.J.; Jiang, C.; Wu, J.C.; Snyder, M.P. Disruption of mesoderm formation during cardiac differentiation due to developmental exposure to 13-cis-retinoic acid. Sci. Rep. 2018, 8, 12960. [Google Scholar] [CrossRef]
- Messina, G.; Biressi, S.; Monteverde, S.; Magli, A.; Cassano, M.; Perani, L.; Roncaglia, E.; Tagliafico, E.; Starnes, L.; Campbell, C.E.; et al. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell 2010, 140, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Antonini, S.; Bonfanti, C.; Monteverde, S.; Vezzali, C.; Tajbakhsh, S.; Cossu, G.; Messina, G. Nfix Regulates Temporal Progression of Muscle Regeneration through Modulation of Myostatin Expression. Cell Rep. 2016, 14, 2238–2249. [Google Scholar] [CrossRef] [Green Version]
- Botella, L.M.; Sanchez-Elsner, T.; Sanz-Rodriguez, F.; Kojima, S.; Shimada, J.; Guerrero-Esteo, M.; Cooreman, M.P.; Ratziu, V.; Langa, C.; Vary, C.P.; et al. Transcriptional activation of endoglin and transforming growth factor-β signaling components by cooperative interaction between Sp1 and KLF6: Their potential role in the response to vascular injury. Blood 2002, 100, 4001–4010. [Google Scholar] [CrossRef]
- Fisch, S.; Gray, S.; Heymans, S.; Haldar, S.M.; Wang, B.; Pfister, O.; Cui, L.; Kumar, A.; Lin, Z.; Sen-Banerjee, S.; et al. Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 7074–7079. [Google Scholar] [CrossRef] [Green Version]
- Lazo, P.A. Functional implications of tetraspanin proteins in cancer biology. Cancer Sci. 2007, 98, 1666–1677. [Google Scholar] [CrossRef]
- Chen, F.; Kook, H.; Milewski, R.; Gitler, A.D.; Lu, M.M.; Li, J.; Nazarian, R.; Schnepp, R.; Jen, K.; Biben, C.; et al. Hop is an unusual homeobox gene that modulates cardiac development. Cell 2002, 110, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Kook, H.; Lepore, J.J.; Gitler, A.D.; Lu, M.M.; Wing-Man Yung, W.; Mackay, J.; Zhou, R.; Ferrari, V.; Gruber, P.; Epstein, J.A. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J. Clin. Investig. 2003, 112, 863–871. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ensembl_ID | Gene_Symbol | p Value | Average_Log2FC | Effect 1 |
|---|---|---|---|---|
| ENSG00000125740 | FOSB | 0.0000015 | −3.257547079 | Down |
| ENSG00000175161 | CADM2 | 0.0000197 | −2.071624331 | Down |
| ENSG00000176390 | CRLF3 | 0.0000218 | −0.658728667 | Down |
| ENSG00000212901 | KRTAP3-1 | 0.0000095 | 1.661253363 | Up |
| ENSG00000256223 | ZNF10 | 0.0000224 | −0.484320108 | Down |
| ENSG00000185022 | MAFF | 0.0000308 | −1.821593264 | Down |
| ENSG00000039537 | C6 | 0.0001451 | 1.089126636 | Up |
| ENSG00000103037 | SETD6 | 0.0001428 | −0.534750906 | Down |
| ENSG00000147257 | GPC3 | 0.0001270 | 1.743008369 | Up |
| ENSG00000159200 | RCAN1 | 0.0001080 | −1.002579722 | Down |
| ENSG00000161905 | ALOX15 | 0.0000903 | 1.430983226 | Up |
| ENSG00000176194 | CIDEA | 0.0001473 | 1.999963173 | Up |
| ENSG00000197446 | CYP2F1 | 0.0000935 | 0.979810226 | Up |
| ENSG00000205085 | GARIN1A | 0.0001299 | −0.984622408 | Down |
| ENSG00000086289 | EPDR1 | 0.0001936 | 1.404957629 | Up |
| ENSG00000198570 | RD3 | 0.0001984 | −1.503858047 | Down |
| ENSG00000062282 | DGAT2 | 0.0002235 | 1.055794141 | Up |
| ENSG00000244682 | FCGR2C | 0.0002228 | 2.694995426 | Up |
| ENSG00000197006 | METTL9 | 0.0002619 | 0.827325535 | Up |
| ENSG00000006652 | IFRD1 | 0.0003909 | −0.881609917 | Down |
| ENSG00000100167 | SEPTIN3 | 0.0003299 | −0.69613844 | Down |
| ENSG00000108654 | DDX5 | 0.0003928 | −0.422221574 | Down |
| ENSG00000160703 | NLRX1 | 0.0003473 | 0.526989002 | Up |
| ENSG00000162892 | IL24 | 0.0003406 | −3.76275497 | Down |
| ENSG00000183648 | NDUFB1 | 0.0003846 | −0.834568054 | Down |
| ENSG00000065361 | ERBB3 | 0.0004149 | −0.734170375 | Down |
| ENSG00000123243 | ITIH5 | 0.0004356 | 0.866063518 | Up |
| ENSG00000026950 | BTN3A1 | 0.0005153 | 0.841120801 | Up |
| ENSG00000048162 | NOP16 | 0.0005381 | −0.967347096 | Down |
| ENSG00000103404 | USP31 | 0.0006926 | −0.587270031 | Down |
| ENSG00000109846 | CRYAB | 0.0006931 | −0.858475283 | Down |
| ENSG00000112164 | GLP1R | 0.0006748 | 1.339457951 | Up |
| ENSG00000114062 | UBE3A | 0.0005701 | −0.504043138 | Down |
| ENSG00000127527 | EPS15L1 | 0.0007000 | 0.412614122 | Up |
| ENSG00000137033 | IL33 | 0.0006977 | 0.738889805 | Up |
| ENSG00000137338 | PGBD1 | 0.0006317 | −0.641698806 | Down |
| ENSG00000153234 | NR4A2 | 0.0005670 | −2.024169309 | Down |
| ENSG00000155893 | PXYLP1 | 0.0004983 | −1.144178122 | Down |
| ENSG00000163444 | TMEM183A | 0.0006250 | −0.468754302 | Down |
| ENSG00000170242 | USP47 | 0.0006670 | −0.643634597 | Down |
| ENSG00000184566 | NONE- | 0.0006732 | 1.200758974 | Up |
| ENSG00000196757 | ZNF700 | 0.0007092 | −0.676810871 | Down |
| ENSG00000142178 | SIK1 | 0.0007473 | −1.098038824 | Down |
| ENSG00000258311 | NONE | 0.0007591 | −1.27119987 | Down |
| ENSG00000105221 | AKT2 | 0.0008704 | 0.344111036 | Up |
| ENSG00000113739 | STC2 | 0.0008295 | −1.35464027 | Down |
| ENSG00000116761 | CTH | 0.0008316 | −1.69781611 | Down |
| ENSG00000128272 | ATF4 | 0.0007886 | −0.998749847 | Down |
| ENSG00000144655 | CSRNP1 | 0.0008495 | −0.983291446 | Down |
| ENSG00000167920 | KRT10-AS1 | 0.0008554 | 0.855157383 | Up |
| Logo | Alt ID 1 | p Value 2 | TP 3 | FP 4 | Enrichment Ratio 5 | Score Threshold 6 | Average_Log2FC in RNA-seq | p Value in RNA-seq | GIC Score 7 |
|---|---|---|---|---|---|---|---|---|---|
 | MA1517.1.KLF6 | 3.19 × 10−7 | 59/107 (55.14%) | 23/107 (21.5%) | 2.5 | 13 | −0.83 | 0.0091 | 0.547 |
 | MA1870.1.KLF7 | 8.01 × 10−4 | 66/107 (61.68%) | 42/107 (39.25%) | 1.56 | 12 | −0.54 | 0.038 | 0.746 |
 | MA0493.2.KLF1 | 1.06 × 10−4 | 102/107 (95.33%) | 83/107 (77.57%) | 1.23 | 10 | −0.81 | 0.026 | 0.541 |
 | MA01119.1. NFIC::TLX1 | 3.39 × 10−4 | 33/107 (30.8%) | 12/107 (11.2%) | 2.62 | 9.31 | −0.30 | 0.058 | 0.79 |
| Gene Name | seq_Score 1 | seq_Class 2 |
|---|---|---|
| SLC19A2 | 17 | tp |
| LEFTY1 | 14.18 | tp |
| BPIFB3 | 13.5 | tp |
| ECEL1 | 13.37 | tp |
| TRIM42 | 13.11 | tp |
| KRT80 | 11.84 | tp |
| REC114 | 11.79 | tp |
| MPP3 | 10.55 | tp |
| NXNL1 | 10.2 | tp |
| ABCC12 | 10.16 | tp |
| CLEC5A | 10.11 | tp |
| TNFRSF11B | 10.09 | tp |
| EIF4E1B | 10.05 | tp |
| REM2 | 10.01 | tp |
| LAD1 | 9.98 | tp |
| NPAS4 | 9.93 | tp |
| KCNE4 | 9.77 | tp |
| EDN2 | 9.7 | tp |
| ABCC3 | 9.6 | tp |
| MYRFL | 9.56 | tp |
| AGXT | 9.4 | tp |
| TSPAN1 | 9.4 | tp |
| MAFF | 9.4 | tp |
| HOPX | 9.4 | tp |
| SPATA9 | 9.4 | tp |
| DMRTB1 | 9.4 | tp |
| SLC6A20 | 9.4 | tp |
| SPHK1 | 9.4 | tp |
| TMPRSS4 | 9.4 | tp |
| FOSB | 9.4 | tp |
| SEL1L2 | 9.4 | tp |
| DRD4 | 9.4 | tp |
| ABRA | 9.4 | tp |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Y.; Jin, Q.; Gong, Q.; Li, A.; Sun, M.; Jiang, S.; Jin, Y.; Zhang, Z.; He, J.; Zhuang, L. Bioinformatics and Experimental Analyses Reveal NFIC as an Upstream Transcriptional Regulator for Ischemic Cardiomyopathy. Genes 2022, 13, 1051. https://doi.org/10.3390/genes13061051
Ye Y, Jin Q, Gong Q, Li A, Sun M, Jiang S, Jin Y, Zhang Z, He J, Zhuang L. Bioinformatics and Experimental Analyses Reveal NFIC as an Upstream Transcriptional Regulator for Ischemic Cardiomyopathy. Genes. 2022; 13(6):1051. https://doi.org/10.3390/genes13061051
Chicago/Turabian StyleYe, Yang, Qiao Jin, Qian Gong, Aoqi Li, Minghao Sun, Sibo Jiang, Yulan Jin, Zhe Zhang, Jin He, and Lenan Zhuang. 2022. "Bioinformatics and Experimental Analyses Reveal NFIC as an Upstream Transcriptional Regulator for Ischemic Cardiomyopathy" Genes 13, no. 6: 1051. https://doi.org/10.3390/genes13061051
APA StyleYe, Y., Jin, Q., Gong, Q., Li, A., Sun, M., Jiang, S., Jin, Y., Zhang, Z., He, J., & Zhuang, L. (2022). Bioinformatics and Experimental Analyses Reveal NFIC as an Upstream Transcriptional Regulator for Ischemic Cardiomyopathy. Genes, 13(6), 1051. https://doi.org/10.3390/genes13061051