
Extension of PERMANOVA to Testing the Mediation Effect of the Microbiome

Abstract
:1. Introduction
2. Materials and Methods
2.1. Motivation toward Inverse Regression
2.2. Overview of PERMANOVA
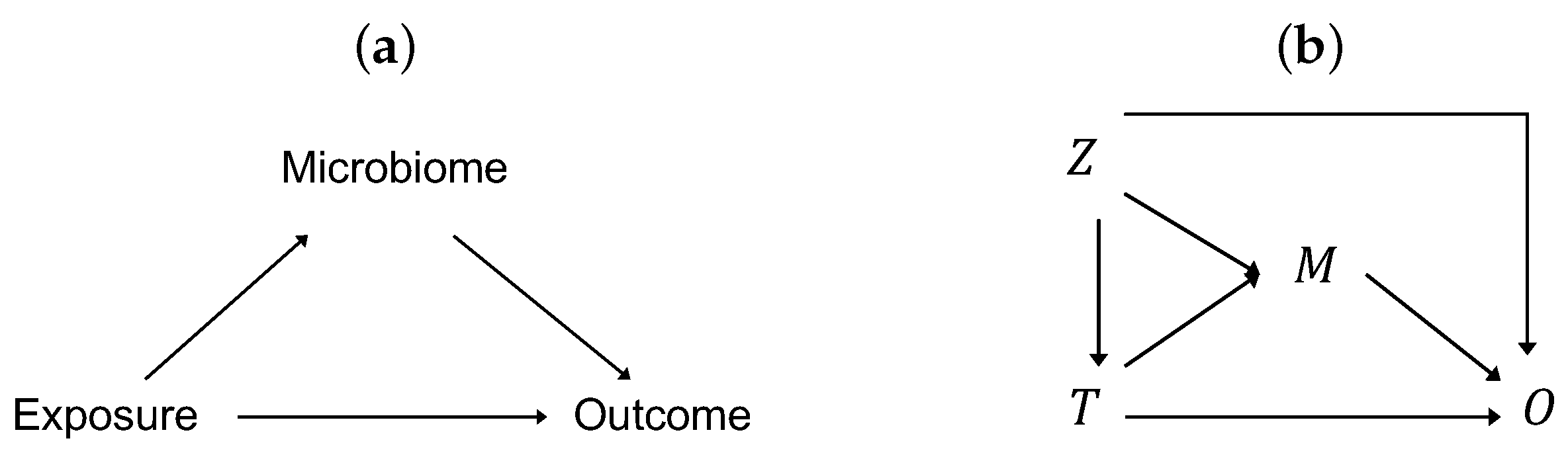
2.3. PERMANOVA-med: Extension of PERMANOVA to Mediation Analysis
2.4. Overview of MedTest and MODIMA
2.5. Availability and Implementation
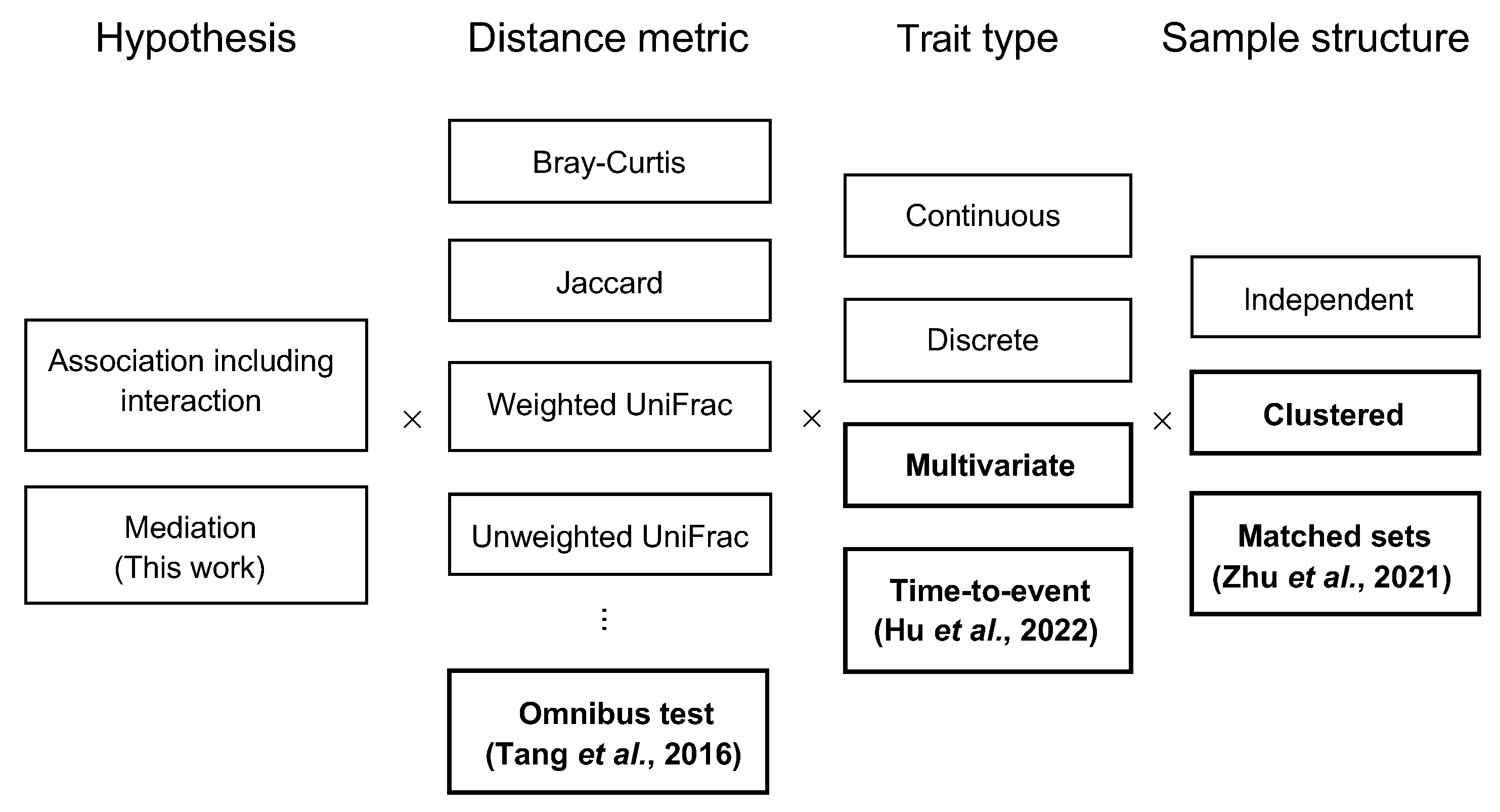
3. Results
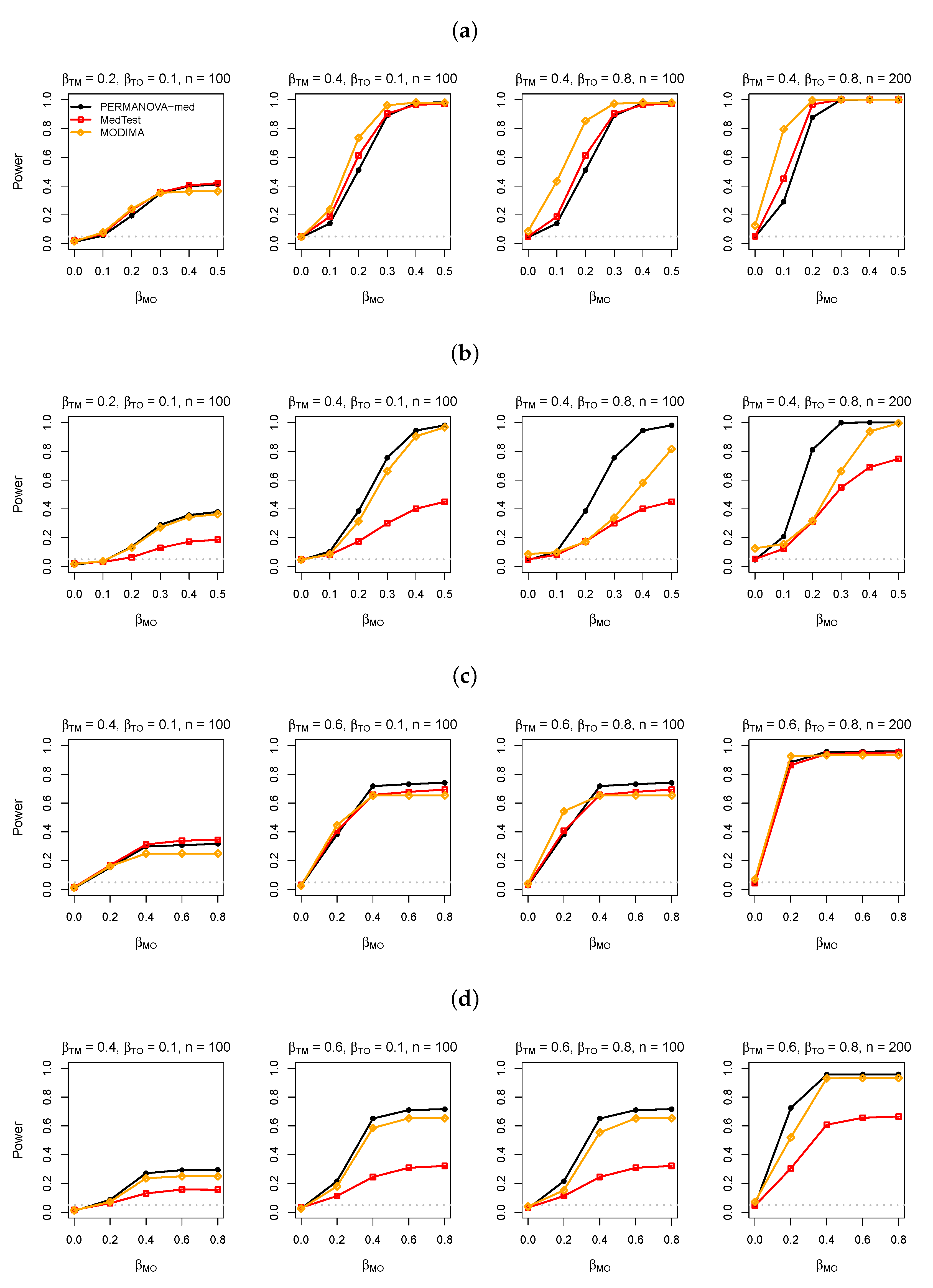
3.1. Simulation Studies
3.2. Simulation Results
3.3. Real Data on Melanoma Immunotherapy Response
3.4. Real Data on Dietary Fiber Intake and BMI
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bai, J.; Hu, Y.; Bruner, D. Composition of gut microbiota and its association with body mass index and lifestyle factors in a cohort of 7–18 years old children from the American Gut Project. Pediatr. Obes. 2019, 14, e12480. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American Gut: An open platform for citizen science microbiome research. Msystems 2018, 3, e00031-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.J.; Satten, G.A. Testing hypotheses about the microbiome using the linear decomposition model (LDM). Bioinformatics 2020, 36, 4106–4115. [Google Scholar] [CrossRef]
- Zhu, Z.; Satten, G.A.; Mitchell, C.; Hu, Y.J. Constraining PERMANOVA and LDM to within-set comparisons by projection improves the efficiency of analyses of matched sets of microbiome data. Microbiome 2021, 9, 1–19. [Google Scholar] [CrossRef]
- Legendre, P.; Anderson, M.J. Distance-based redundancy analysis: Testing multispecies responses in multifactorial ecological experiments. Ecol. Monogr. 1999, 69, 1–24. [Google Scholar] [CrossRef]
- McArdle, B.H.; Anderson, M.J. Fitting multivariate models to community data: A comment on distance-based redundancy analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Zhao, N.; Chen, J.; Carroll, I.M.; Ringel-Kulka, T.; Epstein, M.P.; Zhou, H.; Zhou, J.J.; Ringel, Y.; Li, H.; Wu, M.C. Testing in microbiome-profiling studies with MiRKAT, the microbiome regression-based kernel association test. Am. J. Hum. Genet. 2015, 96, 797–807. [Google Scholar] [CrossRef] [Green Version]
- Alekseyenko, A.V. Multivariate Welch t-test on distances. Bioinformatics 2016, 32, 3552–3558. [Google Scholar]
- Zhang, Y.; Han, S.W.; Cox, L.M.; Li, H. A multivariate distance-based analytic framework for microbial interdependence association test in longitudinal study. Genet. Epidemiol. 2017, 41, 769–778. [Google Scholar] [CrossRef]
- Jaccard, P. The distribution of the flora in the alpine zone. New Phytol. 1912, 11, 37–50. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 326–349. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, Z.; Chen, J. A distance-based approach for testing the mediation effect of the human microbiome. Bioinformatics 2018, 34, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, B.; Wallace, K.; Alekseyenko, A.V. MODIMA, a method for multivariate omnibus distance mediation analysis, allows for integration of multivariate exposure-mediator-response relationships. Genes 2019, 10, 524. [Google Scholar] [CrossRef] [Green Version]
- Székely, G.J.; Rizzo, M.L.; Bakirov, N.K. Measuring and testing dependence by correlation of distances. Ann. Stat. 2007, 35, 2769–2794. [Google Scholar] [CrossRef]
- Székely, G.J.; Rizzo, M.L. Brownian distance covariance. Ann. Appl. Stat. 2009, 3, 1236–1265. [Google Scholar] [CrossRef] [Green Version]
- Székely, G.J.; Rizzo, M.L. Partial distance correlation with methods for dissimilarities. Ann. Stat. 2014, 42, 2382–2412. [Google Scholar] [CrossRef]
- Hu, Y.; Satten, G.A.; Hu, Y.J. Testing microbiome associations with censored survival outcomes at both the community and individual taxon levels. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tang, Z.Z.; Chen, G.; Alekseyenko, A.V. PERMANOVA-S: Association test for microbial community composition that accommodates confounders and multiple distances. Bioinformatics 2016, 32, 2618–2625. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.J.; Satten, G.A. A rarefaction-without-resampling extension of PERMANOVA for testing presence-absence associations in the microbiome. bioRxiv 2021. [Google Scholar] [CrossRef]
- Baron, R.M.; Kenny, D.A. The moderator–mediator variable distinction in social psychological research: Conceptual, strategic, and statistical considerations. J. Personal. Soc. Psychol. 1986, 51, 1173. [Google Scholar] [CrossRef]
- VanderWeele, T.J.; Vansteelandt, S. Conceptual issues concerning mediation, interventions and composition. Stat. Its Interface 2009, 2, 457–468. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, P.F.; Hoggart, C.J.; Pomyen, Y.; Calboli, F.C.; Elliott, P.; Jarvelin, M.R.; Coin, L.J. MultiPhen: Joint model of multiple phenotypes can increase discovery in GWAS. PLoS ONE 2012, 7, e34861. [Google Scholar] [CrossRef]
- Wu, B.; Pankow, J.S. Statistical methods for association tests of multiple continuous traits in genome-wide association studies. Ann. Hum. Genet. 2015, 79, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, A.; Witte, J.S.; Ghosh, S. Semiparametric allelic tests for mapping multiple phenotypes: Binomial regression and Mahalanobis distance. Genet. Epidemiol. 2015, 39, 635–650. [Google Scholar] [CrossRef] [Green Version]
- Gower, J.C. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 1966, 53, 325–338. [Google Scholar] [CrossRef]
- Freedman, D.; Lane, D. A nonstochastic interpretation of reported significance levels. J. Bus. Econ. Stat. 1983, 1, 292–298. [Google Scholar]
- Charlson, E.S.; Chen, J.; Custers-Allen, R.; Bittinger, K.; Li, H.; Sinha, R.; Hwang, J.; Bushman, F.D.; Collman, R.G. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS ONE 2010, 5, e15216. [Google Scholar] [CrossRef] [Green Version]
- Spencer, C.N.; McQuade, J.L.; Gopalakrishnan, V.; McCulloch, J.A.; Vetizou, M.; Cogdill, A.P.; Khan, M.A.W.; Zhang, X.; White, M.G.; Peterson, C.B.; et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 2021, 374, 1632–1640. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Sun, X.; Rajesh, K.; Chalasani, N.; Gelow, K.; Katz, B.; Shah, V.H.; Sanyal, A.J.; Smirnova, E. Effects of rare microbiome taxa filtering on statistical analysis. Front. Microbiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Hu, Y.J. A new approach to testing mediation of the microbiome at both the community and individual taxon levels. Bioinformatics 2022. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scenario | Exposure | n | PERMANOVA-med | MedTest | MODIMA | ||
|---|---|---|---|---|---|---|---|
| M-common | Binary | 0.2 | 0.1 | 100 | 0.012 | 0.021 | 0.017 |
| 0.4 | 0.1 | 100 | 0.044 | 0.049 | 0.046 | ||
| 0.4 | 0.8 | 100 | 0.044 | 0.049 | 0.086 | ||
| 0.4 | 0.8 | 200 | 0.046 | 0.052 | 0.126 | ||
| Continuous | 0.4 | 0.1 | 100 | 0.009 | 0.016 | 0.013 | |
| 0.6 | 0.1 | 100 | 0.026 | 0.032 | 0.025 | ||
| 0.6 | 0.8 | 100 | 0.026 | 0.032 | 0.040 | ||
| 0.6 | 0.8 | 200 | 0.048 | 0.045 | 0.072 | ||
| M-mixed | Binary | 0.4 | 0.1 | 100 | 0.014 | 0.019 | 0.017 |
| 0.6 | 0.1 | 100 | 0.039 | 0.043 | 0.040 | ||
| 0.6 | 0.8 | 100 | 0.039 | 0.043 | 0.047 | ||
| 0.6 | 0.8 | 200 | 0.048 | 0.049 | 0.068 | ||
| Continuous | 0.6 | 0.1 | 100 | 0.004 | 0.010 | 0.007 | |
| 0.8 | 0.1 | 100 | 0.011 | 0.016 | 0.013 | ||
| 0.8 | 0.8 | 100 | 0.011 | 0.016 | 0.016 | ||
| 0.8 | 0.8 | 200 | 0.027 | 0.033 | 0.038 | ||
| M-rare | Binary | 0.2 | 0.1 | 100 | 0.039 | 0.041 | 0.042 |
| 0.4 | 0.1 | 100 | 0.050 | 0.028 | 0.041 | ||
| 0.4 | 0.8 | 100 | 0.050 | 0.028 | 0.088 | ||
| 0.4 | 0.8 | 200 | 0.052 | 0.023 | 0.125 | ||
| Continuous | 0.6 | 0.1 | 100 | 0.045 | 0.046 | 0.042 | |
| 0.8 | 0.1 | 100 | 0.044 | 0.034 | 0.039 | ||
| 0.8 | 0.8 | 100 | 0.044 | 0.034 | 0.082 | ||
| 0.8 | 0.8 | 200 | 0.049 | 0.026 | 0.125 |
| Scenario | PERMANOVA-med | MedTest | MODIMA | |
|---|---|---|---|---|
| M-common | 0.2 | 0.008 | 0.014 | 0.242 |
| 0.4 | 0.035 | 0.040 | 0.385 | |
| M-mixed | 0.4 | 0.006 | 0.015 | 0.056 |
| 0.6 | 0.020 | 0.029 | 0.103 | |
| M-rare | 0.2 | 0.026 | 0.036 | 0.279 |
| 0.4 | 0.046 | 0.035 | 0.238 |
| Outcome | Exposure | PERMANOVA-med | MedTest | MODIMA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | BC | J | Omni | BC | J | Omni | BC | J | Omni | ||
| No adjustment of covariates | |||||||||||
| Progression-free | Fiber intake | 89 | 0.808 | 0.965 | 0.958 | - | - | - | - | - | - |
| survival | Probiotics | 110 | 0.913 | 0.716 | 0.899 | - | - | - | - | - | - |
| Fiber + probiotics (4 levels) | 89 | 0.777 | 0.975 | 0.953 | - | - | - | - | - | - | |
| Sufficient fiber + no probiotics | 89 | 0.717 | 0.965 | 0.910 | - | - | - | - | - | - | |
| Response to ICB | Fiber intake | 94 | 0.727 | 0.955 | 0.903 | 0.624 | 0.636 | 0.837 | 0.384 | 0.935 | - |
| Probiotics | 110 | 0.888 | 0.589 | 0.794 | 0.978 | 0.698 | 0.898 | 0.915 | 0.381 | - | |
| Fiber + probiotics (4 levels) | 89 | 0.620 | 0.980 | 0.827 | - | - | - | 0.430 | 0.947 | - | |
| Sufficient fiber + no probiotics | 89 | 0.490 | 0.955 | 0.697 | 0.276 | 0.626 | 0.455 | 0.441 | 0.947 | - | |
| Adjusting for BMI, prior treatment, LDH, stage | |||||||||||
| Progression-free | Fiber intake | 89 | 0.786 | 0.990 | 0.936 | - | - | - | - | - | - |
| survival | Probiotics | 110 | 0.983 | 0.788 | 0.947 | - | - | - | - | - | - |
| Fiber + probiotics (4 levels) | 89 | 0.770 | 0.995 | 0.935 | - | - | - | - | - | - | |
| Sufficient fiber + no probiotics | 89 | 0.725 | 0.980 | 0.903 | - | - | - | - | - | - | |
| Response to ICB | Fiber intake | 94 | 0.870 | 0.920 | 0.975 | 0.832 | 0.935 | 0.966 | - | - | - |
| Probiotics | 110 | 0.973 | 0.433 | 0.630 | 0.911 | 0.539 | 0.773 | - | - | - | |
| Fiber + probiotics (4 levels) | 89 | 0.760 | 0.975 | 0.928 | - | - | - | - | - | - | |
| Sufficient fiber + no probiotics | 89 | 0.644 | 0.925 | 0.850 | 0.453 | 0.973 | 0.682 | - | - | - | |
| Method | BC | J | UniFrac | WUniFrac | GUniFrac | Omni | |
|---|---|---|---|---|---|---|---|
| No filter | PERMANOVA-med | 0.304 | 0.032 | 0.0490 | 0.597 | 0.235 | 0.0859 |
| MedTest | 0.530 | 0.00400 | 0.0739 | 0.792 | 0.521 | 0.0110 | |
| MODIMA | 0.087 | 0.00800 | 0.0560 | 0.451 | 0.250 | - | |
| With filter | PERMANOVA-med | 0.289 | 0.036 | 0.0769 | 0.586 | 0.252 | 0.0929 |
| MedTest | 0.526 | 0.114 | 0.766 | 0.706 | 0.438 | 0.335 | |
| MODIMA | 0.084 | 0.005 | 0.077 | 0.351 | 0.193 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, Y.; Hu, Y.-J. Extension of PERMANOVA to Testing the Mediation Effect of the Microbiome. Genes 2022, 13, 940. https://doi.org/10.3390/genes13060940
Yue Y, Hu Y-J. Extension of PERMANOVA to Testing the Mediation Effect of the Microbiome. Genes. 2022; 13(6):940. https://doi.org/10.3390/genes13060940
Chicago/Turabian StyleYue, Ye, and Yi-Juan Hu. 2022. "Extension of PERMANOVA to Testing the Mediation Effect of the Microbiome" Genes 13, no. 6: 940. https://doi.org/10.3390/genes13060940
APA StyleYue, Y., & Hu, Y. -J. (2022). Extension of PERMANOVA to Testing the Mediation Effect of the Microbiome. Genes, 13(6), 940. https://doi.org/10.3390/genes13060940