
Construction and Validation of a Tumor Microenvironment-Based Scoring System to Evaluate Prognosis and Response to Immune Checkpoint Inhibitor Therapy in Lung Adenocarcinoma Patients

Abstract
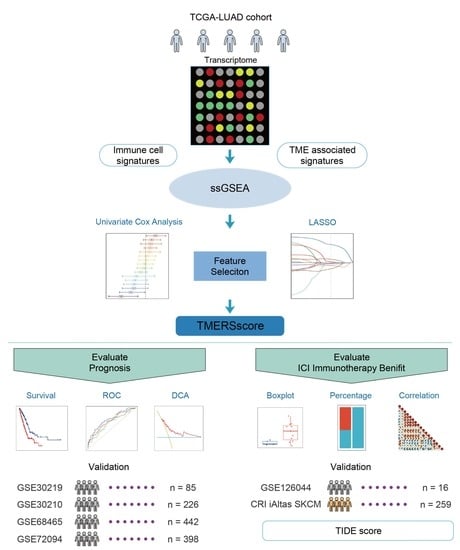
:
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Preprocessing
2.2. Collection and Quantification of Tumor Microenvironment-Related Signatures
2.3. Establishment of the Tumor Microenvironment-Related Signature Score
2.4. Comparison of Tumor Microenvironment-Related Signature Score with Other Prognostic Models
2.5. Identification of Differentially Expressed Genes
2.6. Bioinformatics Analysis of Differentially Expressed Genes
2.7. Evaluation of ICI Treatment Response by Tumor Microenvironment-Related Signature Score
2.8. Statistical Analysis
3. Results
3.1. Construction of a Tumor Microenvironment-Related Signature Score for Significant Stratification of LUAD Patients
3.2. Validation of Tumor Microenvironment-Related Signature Score in Predicting Prognosis of LUAD Patients
3.3. Revelation of the Underlying Reasons behind Tumor Microenvironment-Related Signature Score
3.4. Evaluation of Tumor Malignancy and ICI Immunotherapy Efficacy by Tumor Microenvironment-Related Signature Score
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bade, B.C.; Dela Cruz, C.S. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020, 41, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, T.V.; Budkevich, I.N.; Zhivotovsky, B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018, 9, 117. [Google Scholar] [CrossRef]
- Li, L.; Sun, Y.; Feng, M.; Wang, L.; Liu, J. Clinical significance of blood-based miRNAs as biomarkers of non-small cell lung cancer. Oncol. Lett. 2018, 15, 8915–8925. [Google Scholar] [CrossRef] [Green Version]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.; Chirieac, L.R.; D’Amico, T.A.; DeCamp, M.M.; Dilling, T.J.; Dobelbower, M.; et al. Non-Small Cell Lung Cancer, Version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 504–535. [Google Scholar] [CrossRef] [PubMed]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Ghoshdastider, U.; Rohatgi, N.; Mojtabavi Naeini, M.; Baruah, P.; Revkov, E.; Guo, Y.A.; Rizzetto, S.; Wong, A.M.L.; Solai, S.; Nguyen, T.T.; et al. Pan-Cancer Analysis of Ligand-Receptor Cross-talk in the Tumor Microenvironment. Cancer Res. 2021, 81, 1802–1812. [Google Scholar] [CrossRef]
- Korneev, K.V.; Atretkhany, K.N.; Drutskaya, M.S.; Grivennikov, S.I.; Kuprash, D.V.; Nedospasov, S.A. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine 2017, 89, 127–135. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef] [Green Version]
- Karachaliou, N.; Gonzalez-Cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Gimenez-Capitan, A.; Teixido, C.; Molina-Vila, M.A.; Viteri, S.; De Los Llanos Gil, M.; et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther. Adv. Med. Oncol. 2018, 10, 1758834017749748. [Google Scholar] [CrossRef]
- Altundag, O.; Altundag, K.; Morandi, P.; Gunduz, M. Cytokines and chemokines as predictive markers in non-small cell lung cancer patients with brain metastases. Lung Cancer 2005, 47, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Z.; Bao, S.; Yan, C.; Hou, P.; Wu, N.; Su, J.; Xu, L.; Zhou, M. Identification of tumor immune infiltration-associated lncRNAs for improving prognosis and immunotherapy response of patients with non-small cell lung cancer. J. Immunother. Cancer 2020, 8, e000110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Qi, C.; Qin, B.; Kang, X.; Hu, Y.; Han, W. Immune-Stromal Score Signature: Novel Prognostic Tool of the Tumor Microenvironment in Lung Adenocarcinoma. Front. Oncol. 2020, 10, 541330. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, M.; Yoshida, T.; Matsumoto, Y.; Shinno, Y.; Okuma, Y.; Goto, Y.; Horinouchi, H.; Yamamoto, N.; Watanabe, S.I.; Ohe, Y.; et al. Impact of chemoradiotherapy on the immune-related tumour microenvironment and efficacy of anti-PD-(L)1 therapy for recurrences after chemoradiotherapy in patients with unresectable locally advanced non-small cell lung cancer. Eur. J. Cancer 2020, 140, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Iams, W.; Cruz, M.R.; Chandra, S.; Choi, J.; Giles, F. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2018, 6, 39. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Sanmamed, M.F.; Hastings, K.; Politi, K.; Rimm, D.L.; Chen, L.; Melero, I.; Schalper, K.A.; Herbst, R.S. Immunotherapy in Non-Small Cell Lung Cancer: Facts and Hopes. Clin. Cancer Res. 2019, 25, 4592–4602. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Shalabi, A.; Hubbard-Lucey, V.M. Comprehensive analysis of the clinical immuno-oncology landscape. Ann. Oncol. 2018, 29, 84–91. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Marshall, H.T.; Djamgoz, M.B.A. Immuno-Oncology: Emerging Targets and Combination Therapies. Front. Oncol. 2018, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Diab, A.; Abdallah, K.; Bingham, C.O., 3rd; Brogdon, C.; Dadu, R.; Hamad, L.; Kim, S.; Lacouture, M.E.; LeBoeuf, N.R.; et al. Managing toxicities associated with immune checkpoint inhibitors: Consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J. Immunother. Cancer 2017, 5, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, J.A.; Thorsson, V.; Lamb, A.E.; Gibbs, D.L.; Heimann, C.; Yu, J.X.; Chung, V.; Chae, Y.; Dang, K.; Vincent, B.G.; et al. CRI iAtlas: An interactive portal for immuno-oncology research. F1000Research 2020, 9, 1028. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Wang, S.; Xiong, Y.; Zhang, Q.; Su, D.; Yu, C.; Cao, Y.; Pan, Y.; Lu, Q.; Zuo, Y.; Yang, L. Clinical significance and immunogenomic landscape analyses of the immune cell signature based prognostic model for patients with breast cancer. Brief. Bioinform. 2021, 22, bbaa311. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.M.; Lenburg, M.E.; Yau, C.; Boudreau, A.; van ‘t Veer, L.J. Gene co-expression modules as clinically relevant hallmarks of breast cancer diversity. PLoS ONE 2014, 9, e88309. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Andorf, S.; Gomes, L.; Dunn, P.; Schaefer, H.; Pontius, J.; Berger, P.; Desborough, V.; Smith, T.; Campbell, J.; et al. ImmPort: Disseminating data to the public for the future of immunology. Immunol. Res. 2014, 58, 234–239. [Google Scholar] [CrossRef]
- Miao, Y.R.; Zhang, Q.; Lei, Q.; Luo, M.; Xie, G.Y.; Wang, H.; Guo, A.Y. ImmuCellAI: A Unique Method for Comprehensive T-Cell Subsets Abundance Prediction and its Application in Cancer Immunotherapy. Adv. Sci. 2020, 7, 1902880. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Nirmal, A.J.; Regan, T.; Shih, B.B.; Hume, D.A.; Sims, A.H.; Freeman, T.C. Immune Cell Gene Signatures for Profiling the Microenvironment of Solid Tumors. Cancer Immunol. Res. 2018, 6, 1388–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, D.; Ye, Z.; Shen, R.; Yu, G.; Wu, J.; Xiong, Y.; Zhou, R.; Qiu, W.; Huang, N.; Sun, L.; et al. IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front. Immunol. 2021, 12, 687975. [Google Scholar] [CrossRef] [PubMed]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Jian, X.; Liu, Z.; Zhao, J.; Zhang, S.; Lin, Y.; Xie, L. Construction and Validation of an Immune Cell Signature Score to Evaluate Prognosis and Therapeutic Efficacy in Hepatocellular Carcinoma. Front. Genet. 2021, 12, 741226. [Google Scholar] [CrossRef]
- He, L.; Chen, J.; Xu, F.; Li, J.; Li, J. Prognostic Implication of a Metabolism-Associated Gene Signature in Lung Adenocarcinoma. Mol. Ther. Oncol. 2020, 19, 265–277. [Google Scholar] [CrossRef]
- Liu, L.; He, H.; Peng, Y.; Yang, Z.; Gao, S. A four-gene prognostic signature for predicting the overall survival of patients with lung adenocarcinoma. PeerJ 2021, 9, e11911. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Liang, J.; Liu, J.; Tian, D.; Chen, Z. Establishment and validation of an eight-gene metabolic-related prognostic signature model for lung adenocarcinoma. Aging (Albany NY) 2021, 13, 8688–8705. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Jang, S.J.; Khuri, F.R.; Hassan, K.; Liu, D.; Hong, W.K.; Mao, L. Overexpression of cyclin B1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res 2000, 60, 4000–4004. Available online: https://www.ncbi.nlm.nih.gov/pubmed/10945597 (accessed on 15 November 2021). [PubMed]
- Egloff, A.M.; Weissfeld, J.; Land, S.R.; Finn, O.J. Evaluation of anticyclin B1 serum antibody as a diagnostic and prognostic biomarker for lung cancer. Ann. N. Y. Acad. Sci. 2005, 1062, 29–40. [Google Scholar] [CrossRef]
- Ma, C.; Luo, H.; Cao, J.; Gao, C.; Fa, X.; Wang, G. Independent prognostic implications of RRM2 in lung adenocarcinoma. J. Cancer 2020, 11, 7009–7022. [Google Scholar] [CrossRef]
- Wang, J.; He, Z.; Duan, R. Expression of ASPM in Lung Adenocarcinoma and Its Relationship with Development and Prognosis. Zhongguo Fei Ai Za Zhi 2020, 23, 29–35. [Google Scholar] [CrossRef]
- Guo, W.; Sun, S.; Guo, L.; Song, P.; Xue, X.; Zhang, H.; Zhang, G.; Wang, Z.; Qiu, B.; Tan, F.; et al. Elevated TOP2A and UBE2C expressions correlate with poor prognosis in patients with surgically resected lung adenocarcinoma: A study based on immunohistochemical analysis and bioinformatics. J. Cancer Res. Clin. Oncol. 2020, 146, 821–841. [Google Scholar] [CrossRef]
- Scheuermann, R.H.; Racila, E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk. Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef]
- Julamanee, J.; Terakura, S.; Umemura, K.; Adachi, Y.; Miyao, K.; Okuno, S.; Takagi, E.; Sakai, T.; Koyama, D.; Goto, T.; et al. Composite CD79A/CD40 co-stimulatory endodomain enhances CD19CAR-T cell proliferation and survival. Mol. Ther. 2021, 29, 2677–2690. [Google Scholar] [CrossRef]
- Tedder, T.F.; Streuli, M.; Schlossman, S.F.; Saito, H. Isolation and structure of a cDNA encoding the B1 (CD20) cell-surface antigen of human B lymphocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Perovanovic, J.; Shakya, A.; Shen, Z.; German, C.N.; Ibarra, A.; Jafek, J.L.; Lin, N.P.; Evavold, B.D.; Chou, D.H.; et al. Targeting transcriptional coregulator OCA-B/Pou2af1 blocks activated autoreactive T cells in the pancreas and type 1 diabetes. J. Exp. Med. 2021, 218, e20200533. [Google Scholar] [CrossRef] [PubMed]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V.; et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xiao, Q.; Zhou, B.; Dai, Z.; Kang, Y. Prognostic Significance of Programmed Death Ligand 1 Expression and Tumor-Infiltrating Lymphocytes in Axial Osteosarcoma. World Neurosurg. 2019, 129, e240–e254. [Google Scholar] [CrossRef]
- Robinson, B.D.; Sica, G.L.; Liu, Y.F.; Rohan, T.E.; Gertler, F.B.; Condeelis, J.S.; Jones, J.G. Tumor microenvironment of metastasis in human breast carcinoma: A potential prognostic marker linked to hematogenous dissemination. Clin. Cancer Res. 2009, 15, 2433–2441. [Google Scholar] [CrossRef] [Green Version]
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat. Rev. 2014, 40, 558–566. [Google Scholar] [CrossRef]
- Budczies, J.; Kirchner, M.; Kluck, K.; Kazdal, D.; Glade, J.; Allgauer, M.; Kriegsmann, M.; Heussel, C.P.; Herth, F.J.; Winter, H.; et al. A gene expression signature associated with B cells predicts benefit from immune checkpoint blockade in lung adenocarcinoma. Oncoimmunology 2021, 10, 1860586. [Google Scholar] [CrossRef]
- Zhang, Y.; Gallastegui, N.; Rosenblatt, J.D. Regulatory B cells in anti-tumor immunity. Int. Immunol. 2015, 27, 521–530. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reynies, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougouin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Mehnert, J.M.; Monjazeb, A.M.; Beerthuijzen, J.M.T.; Collyar, D.; Rubinstein, L.; Harris, L.N. The Challenge for Development of Valuable Immuno-oncology Biomarkers. Clin. Cancer Res. 2017, 23, 4970–4979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojas-Krawczyk, K.; Kalinka, E.; Grenda, A.; Krawczyk, P.; Milanowski, J. Beyond PD-L1 Markers for Lung Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 1915. [Google Scholar] [CrossRef] [Green Version]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Cohort | ||||
|---|---|---|---|---|---|
| Train | Validation | ||||
| TCGA | GSE30219 | GSE30210 | GSE68465 | GSE72094 | |
| ALL | 479 | 85 | 226 | 442 | 398 |
| Age, average (standard deviation) | 65.2 (10.1) | 61.5 (9.28) | 59.6 (7.40) | 64.4 (10.1) | 69.4 (9.45) |
| Gender: | |||||
| female, % | 254 (53.0%) | 19 (22.4%) | 121 (53.5%) | 219 (49.5%) | 222 (55.8%) |
| male, % | 225 (47.0%) | 66 (77.6%) | 105 (46.5%) | 223 (50.5%) | 176 (44.2%) |
| AJCC pTNM Stage: | |||||
| I, % | 264 (55.1%) | not reported | 168 (74.3%) | not reported | 254 (63.8%) |
| II, % | 118 (24.6%) | not reported | 58 (25.7%) | not reported | 67 (16.8%) |
| III, % | 76 (15.9%) | not reported | 0 (0%) | not reported | 57 (14.3%) |
| IV, % | 21 (4.38%) | not reported | 0 (0%) | not reported | 15 (3.77%) |
| unknown | 0 (0%) | not reported | 0 (0%) | not reported | 5 (1.26%) |
| M stage: | |||||
| M0, % | 321 (67.0%) | 85 (100%) | not reported | not reported | not reported |
| M1, % | 21 (4.38%) | 0 (0%) | not reported | not reported | not reported |
| MX, % | 137 (28.6%) | 0 (0%) | not reported | not reported | not reported |
| T stage: | |||||
| T1, % | 163 (34.0%) | 71 (83.5%) | not reported | 150 (33.9%) | not reported |
| T2, % | 255 (53.2%) | 12 (14.1%) | not reported | 251 (56.8%) | not reported |
| T3, % | 45 (9.39%) | 2 (2.35%) | not reported | 28 (6.33%) | not reported |
| T4, % | 16 (3.34%) | 0 (0%) | not reported | 11 (2.49%) | not reported |
| unknown | 0 (0%) | 0 (0%) | not reported | 2 (0.45%) | not reported |
| N stage: | |||||
| N0, % | 322 (67.2%) | 82 (96.5%) | not reported | 299 (67.6%) | not reported |
| N1, % | 90 (18.8%) | 3 (3.53%) | not reported | 87 (19.7%) | not reported |
| N2, % | 67 (14.0%) | 0 (0%) | not reported | 53 (12.0%) | not reported |
| NX, % | 0 (0%) | 0 (0%) | not reported | 3 (0.68%) | not reported |
| Signatures | C-Index | p-Value |
|---|---|---|
| TMERSscore | 0.6719824 | 3.231601 × 10−13 |
| Lei_Liu_2021 | 0.6719603 | 1.812015 × 10−12 |
| Lulu_He_2020 | 0.6653507 | 8.032247 × 10−13 |
| Weishuang_Ma_2021 | 0.6617354 | 2.155125 × 10−11 |
| age | 0.5110899 | 0.6784349 |
| gender | 0.5315841 | 0.1449488 |
| AJCC pTNM Stage | 0.6437586 | 1.533862 × 10−13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, P.; Xu, L.; Jin, M.; Li, L.; Ke, Y.; Zhang, M.; Zhang, K.; Lu, K.; Huang, G. Construction and Validation of a Tumor Microenvironment-Based Scoring System to Evaluate Prognosis and Response to Immune Checkpoint Inhibitor Therapy in Lung Adenocarcinoma Patients. Genes 2022, 13, 951. https://doi.org/10.3390/genes13060951
Huang P, Xu L, Jin M, Li L, Ke Y, Zhang M, Zhang K, Lu K, Huang G. Construction and Validation of a Tumor Microenvironment-Based Scoring System to Evaluate Prognosis and Response to Immune Checkpoint Inhibitor Therapy in Lung Adenocarcinoma Patients. Genes. 2022; 13(6):951. https://doi.org/10.3390/genes13060951
Chicago/Turabian StyleHuang, Pinzheng, Linfeng Xu, Mingming Jin, Lixi Li, Yizhong Ke, Min Zhang, Kairui Zhang, Kongyao Lu, and Gang Huang. 2022. "Construction and Validation of a Tumor Microenvironment-Based Scoring System to Evaluate Prognosis and Response to Immune Checkpoint Inhibitor Therapy in Lung Adenocarcinoma Patients" Genes 13, no. 6: 951. https://doi.org/10.3390/genes13060951
APA StyleHuang, P., Xu, L., Jin, M., Li, L., Ke, Y., Zhang, M., Zhang, K., Lu, K., & Huang, G. (2022). Construction and Validation of a Tumor Microenvironment-Based Scoring System to Evaluate Prognosis and Response to Immune Checkpoint Inhibitor Therapy in Lung Adenocarcinoma Patients. Genes, 13(6), 951. https://doi.org/10.3390/genes13060951