
Mapping-by-Sequencing Reveals Genomic Regions Associated with Seed Quality Parameters in Brassica napus

 ,
,  ,
, 
 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Material and Trait Measurement
2.2. DNA Extraction and Pooling
2.3. Mapping and Variant Calling
2.4. Generation of the “Gold Standard”for SNV Filtering
2.5. Filter Raw Variants per Pool for Delta Allele Frequency (dAF) Calculation
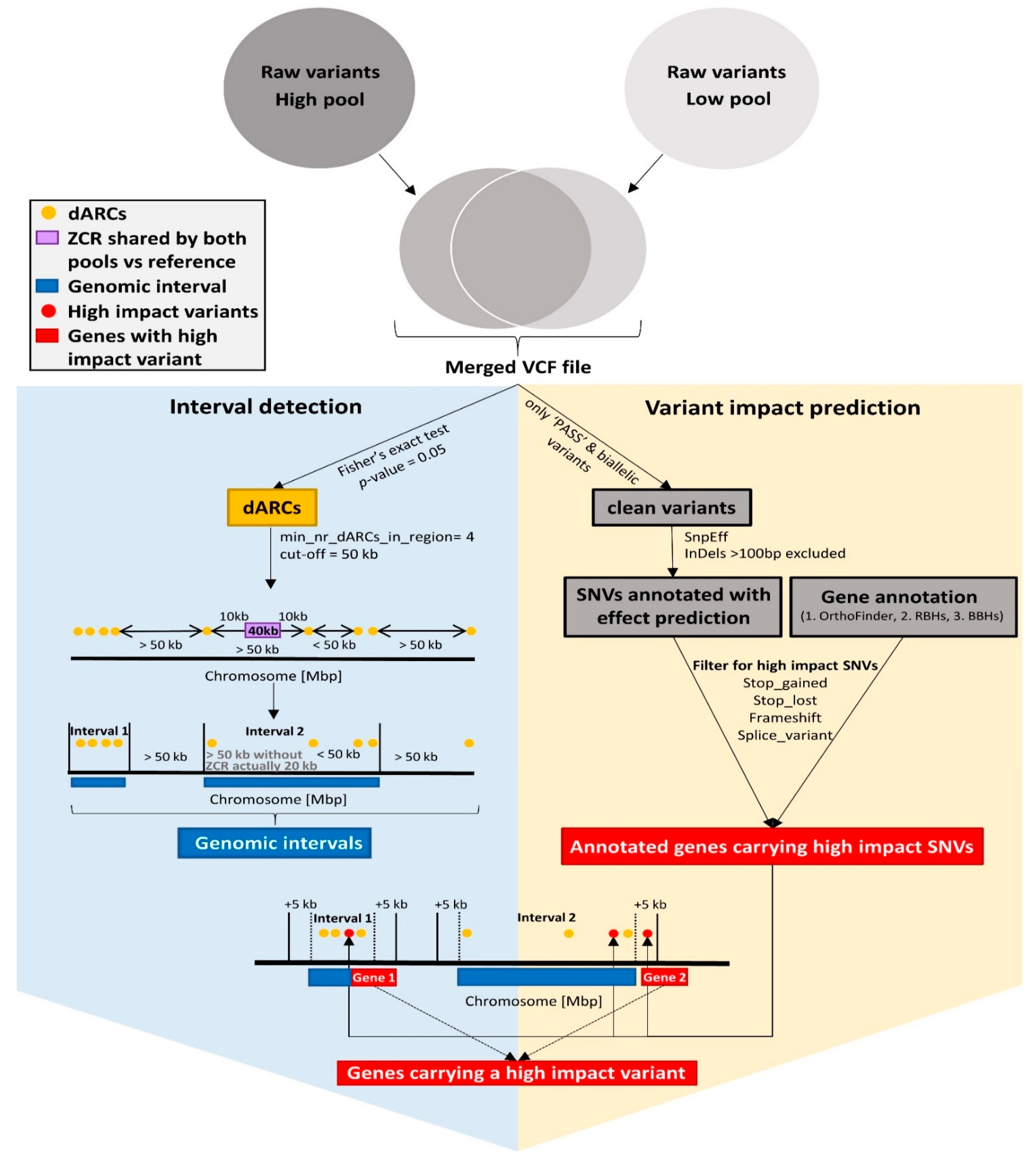
2.6. Interval Detection
2.7. Generation of dAF Plots
2.8. Presence–Absence Variations (PAVs)
2.9. Functional Annotation and Candidate Genes
2.10. Variant Impact Prediction via SnpEff
2.11. Generation and Analysis of RNA-Seq Data
2.12. Identification of MYB Homologs
3. Results
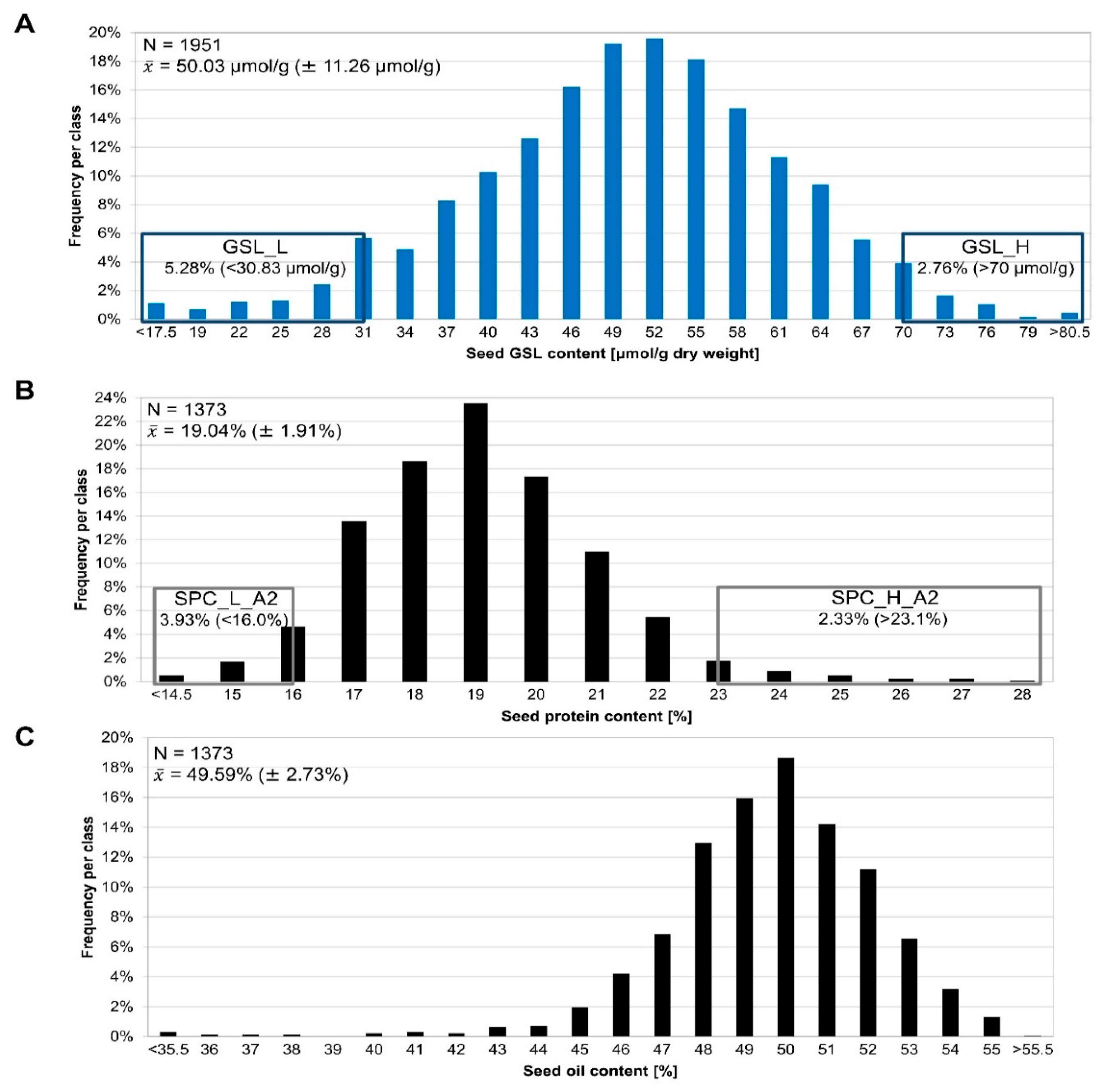
3.1. Phenotyping of the Segregating F2 Population
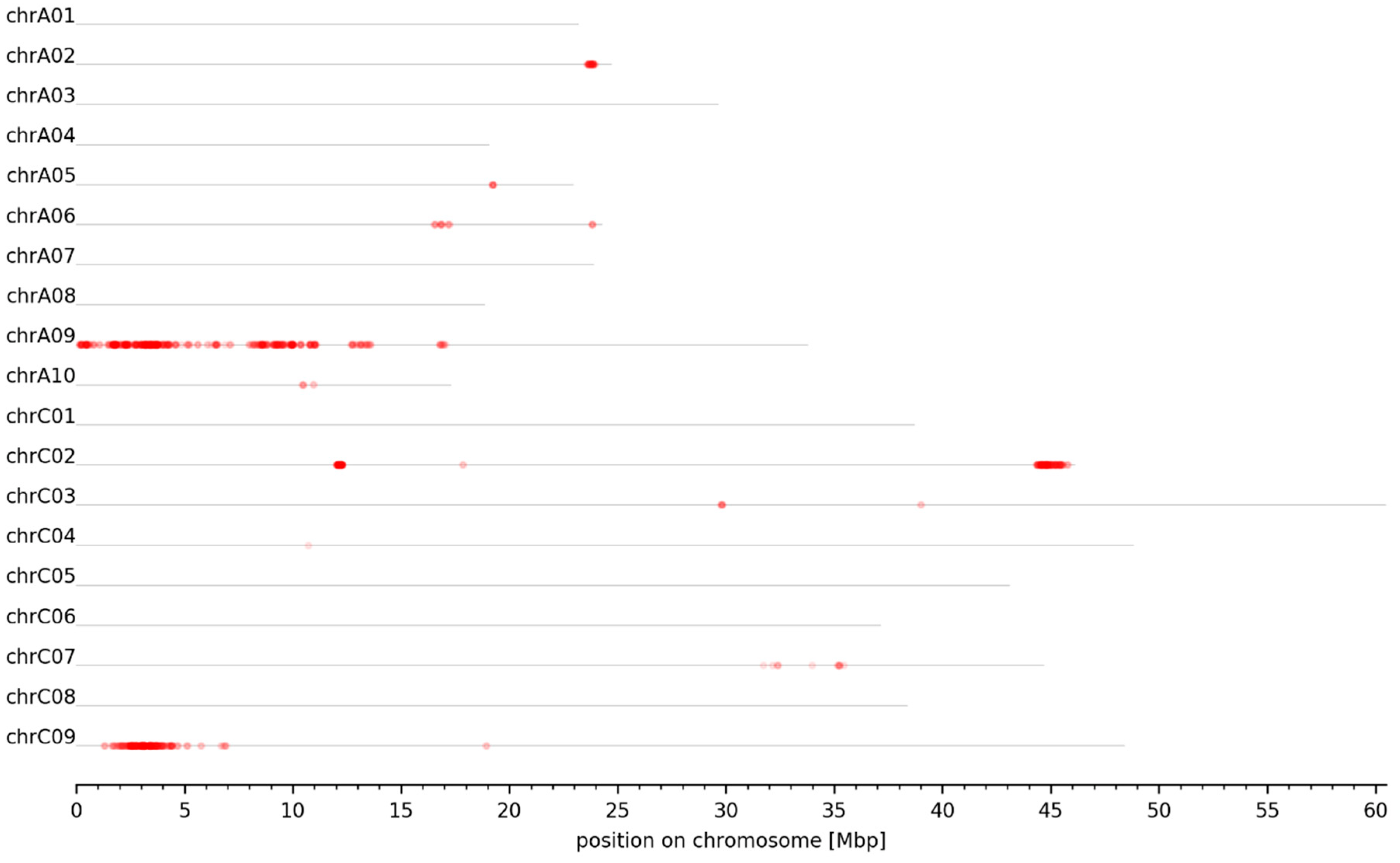
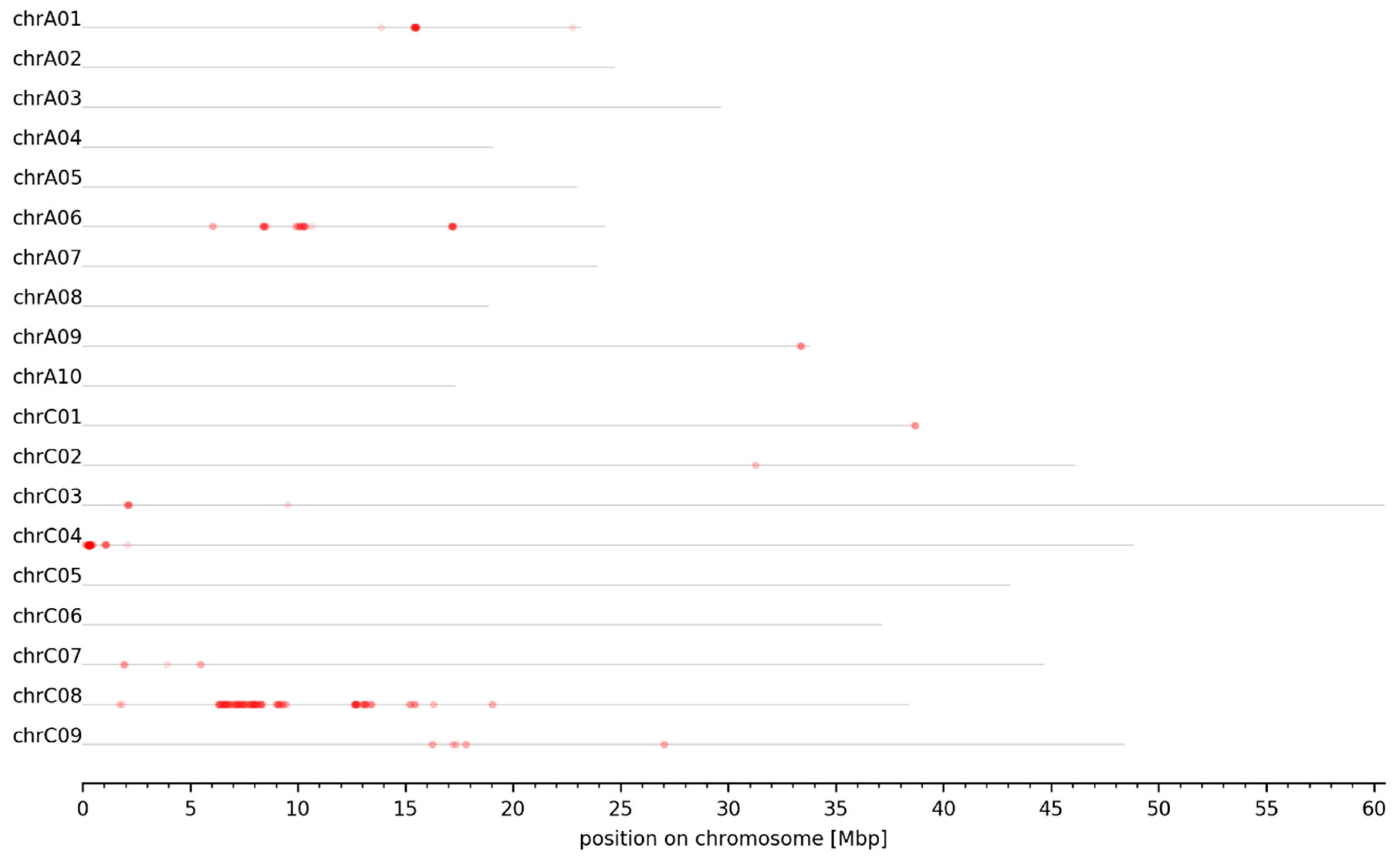
3.2. MBS Predicted Candidate Genomic Intervals Controlling SPC, SOC, and Seed GSL Content—Mapping and Variant Calling
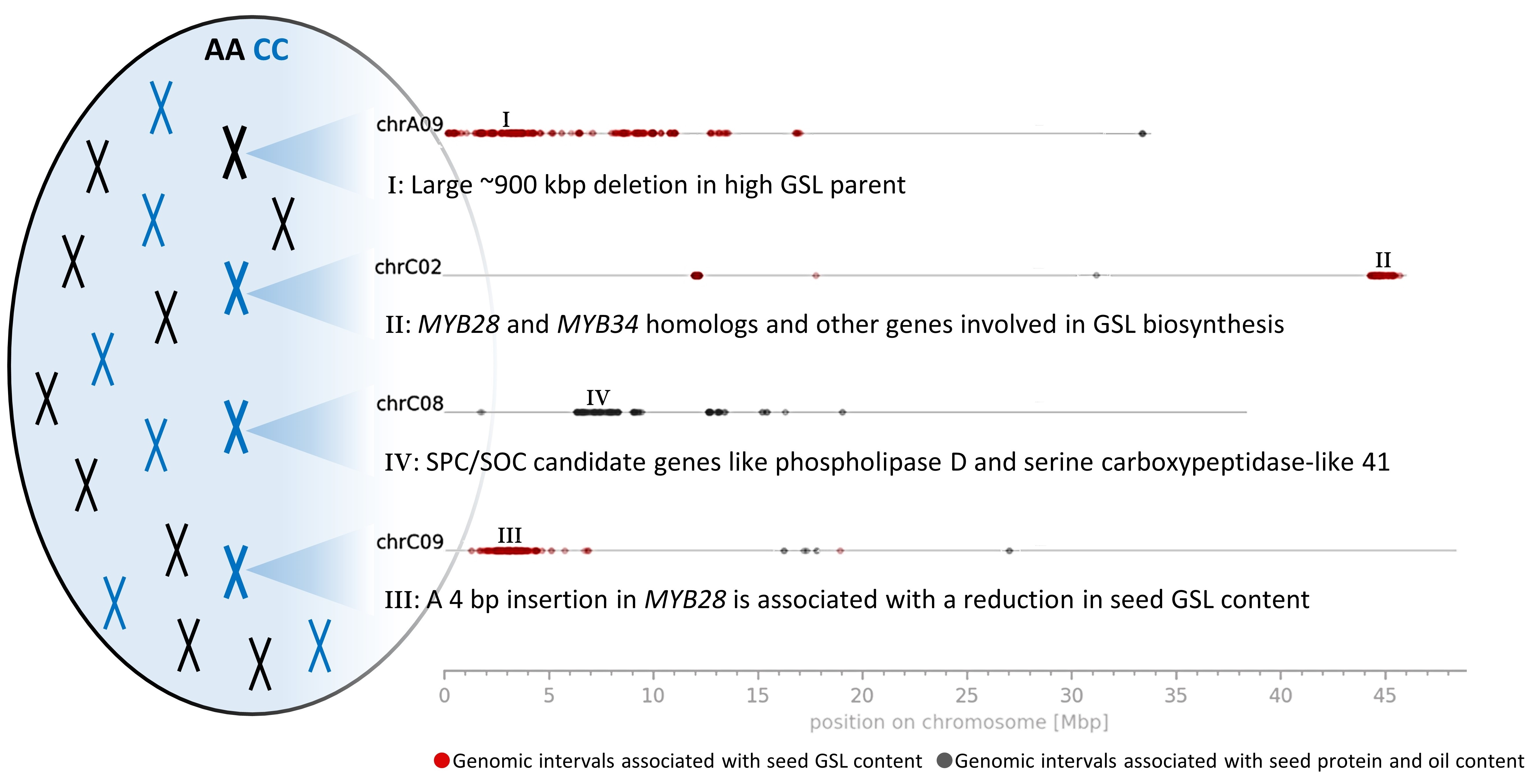
3.3. Genomic Intervals and Candidate Genes Associated with Seed Glucosinolate Content
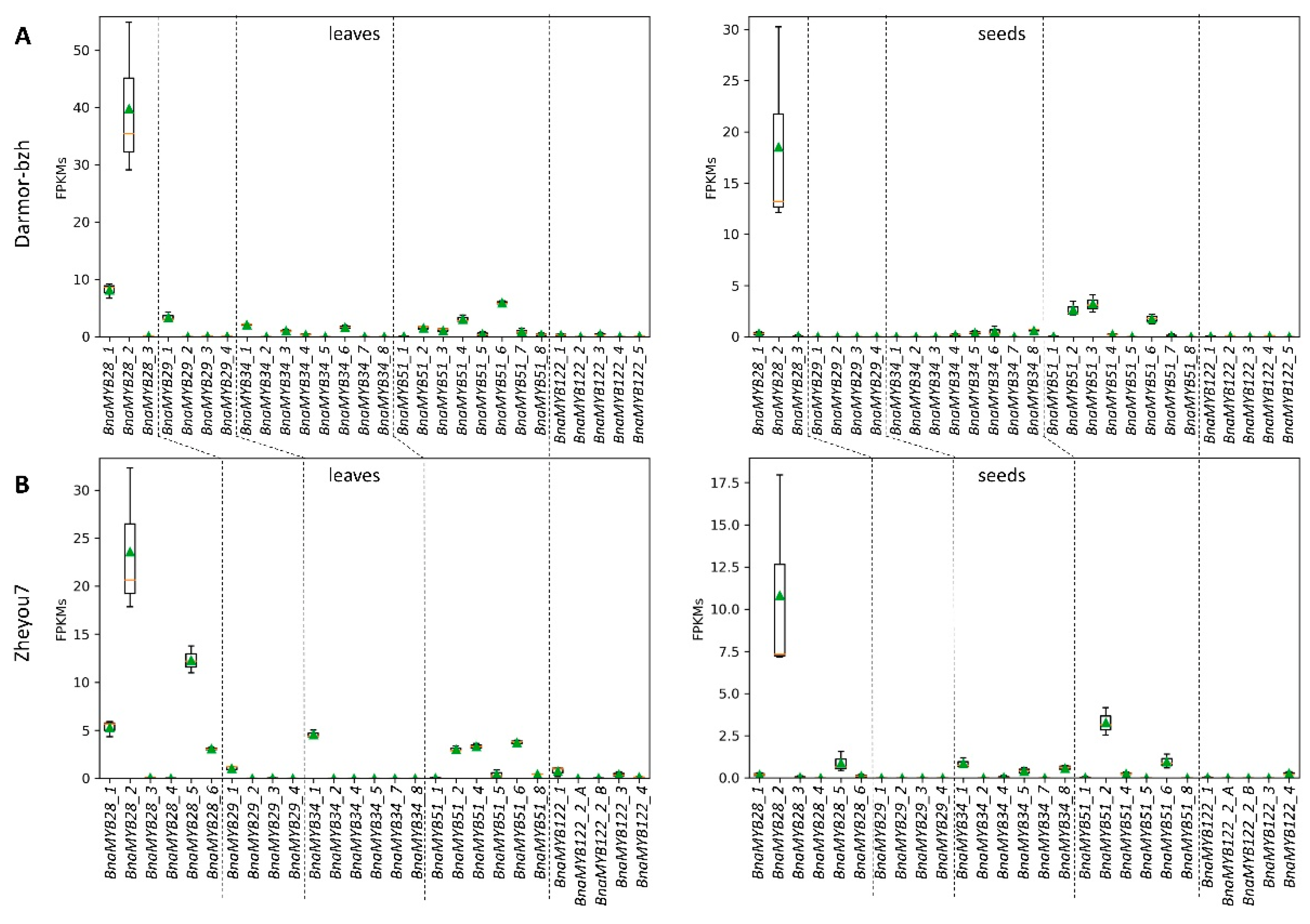
3.3.1. Glucosinolate-Associated MYB Genes Contributed by P1 and P2
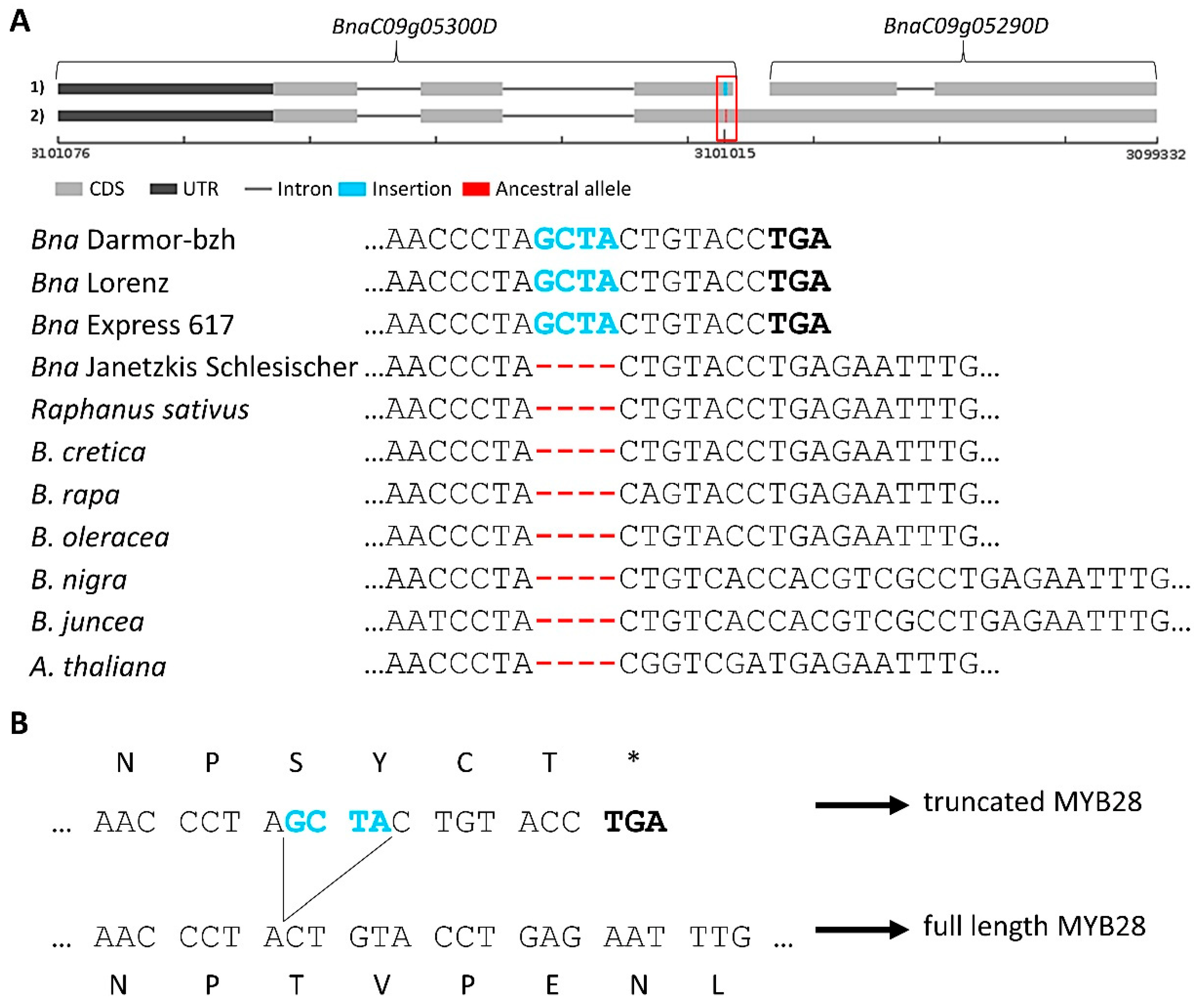
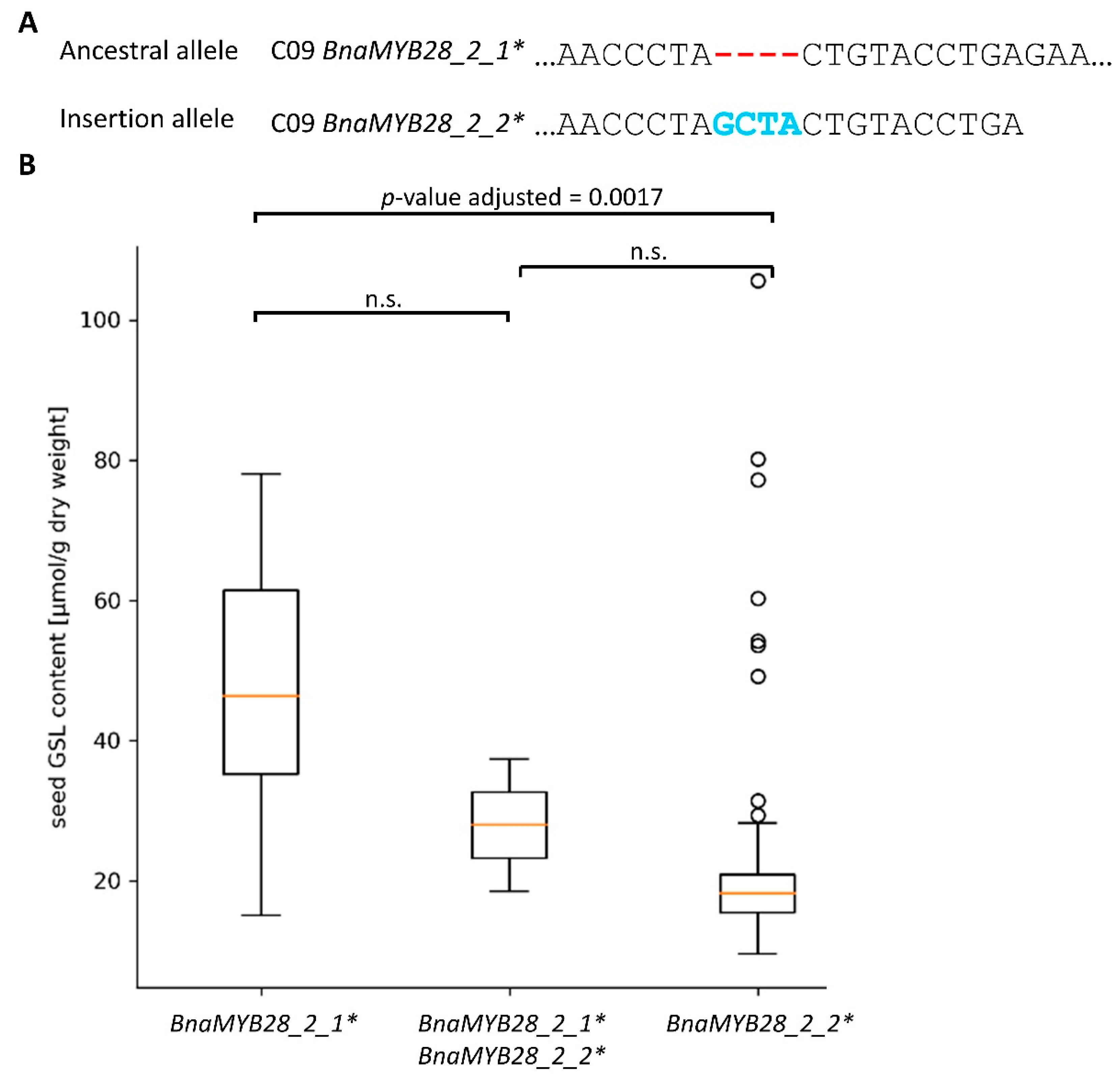
3.3.2. Variation Effects in Genes Involved in Seed Glucosinolate Biosynthesis
3.3.3. PAVs
3.4. Genomic Intervals, Candidate Genes and Variation Effects Associated with Seed Protein and Oil Content
4. Discussion
4.1. Seed Oil and Protein Content
4.2. Seed Glucosinolate Content
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- OECD-FAO; Connell, M.A. OECD-FAO Agricultural Outlook 2015–2024. 2015. Available online: https://www.oecd-ilibrary.org/agriculture-and-food/oecd-fao-agricultural-outlook-2015_agr_outlook-2015-en (accessed on 11 December 2020).
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanasundara, J.P.D.; McIntosh, T.C.; Perera, S.P.; Withana-Gamage, T.S.; Mitra, P. Canola/rapeseed protein-functionality and nutrition. Oilseeds Fats Crops Lipids 2016, 23, D407. [Google Scholar] [CrossRef] [Green Version]
- Nesi, N.; Delourme, R.; Bregeon, M.; Falentin, C.; Renard, M. Genetic and molecular approaches to improve nutritional value of Brassica napus L. seed. Comptes Rendus Biol. 2008, 331, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Fleddermann, M.; Fechner, A.; Rossler, A.; Bahr, M.; Pastor, A.; Liebert, F.; Jahreis, G. Nutritional evaluation of rapeseed protein compared to soy protein for quality, plasma amino acids, and nitrogen balance--a randomized cross-over intervention study in humans. Clin. Nutr. 2013, 32, 519–526. [Google Scholar] [CrossRef]
- Aider, M.; Barbana, C. Canola proteins: Composition, extraction, functional properties, bioactivity, applications as a food ingredient and allergenicity—A practical and critical review. Trends Food Sci. Technol. 2011, 22, 21–39. [Google Scholar] [CrossRef]
- Sonderby, I.E.; Geu-Flores, F.; Halkier, B.A. Biosynthesis of glucosinolates--gene discovery and beyond. Trends Plant Sci. 2010, 15, 283–290. [Google Scholar] [CrossRef]
- Brader, G.; Mikkelsen, M.D.; Halkier, B.A.; Tapio Palva, E. Altering glucosinolate profiles modulates disease resistance in plants. Plant J. 2006, 46, 758–767. [Google Scholar] [CrossRef]
- Barth, C.; Jander, G. Arabidopsis myrosinases TGG1 and TGG2 have redundant function in glucosinolate breakdown and insect defense. Plant J. 2006, 46, 549–562. [Google Scholar] [CrossRef]
- Bednarek, P.; Pislewska-Bednarek, M.; Svatos, A.; Schneider, B.; Doubsky, J.; Mansurova, M.; Humphry, M.; Consonni, C.; Panstruga, R.; Sanchez-Vallet, A.; et al. A glucosinolate metabolism pathway in living plant cells mediates broad-spectrum antifungal defense. Science 2009, 323, 101–106. [Google Scholar] [CrossRef]
- Sotelo, T.; Lema, M.; Soengas, P.; Cartea, M.E.; Velasco, P. In vitro activity of glucosinolates and their degradation products against brassica-pathogenic bacteria and fungi. Appl. Environ. Microbiol. 2015, 81, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Halkier, B.A.; Gershenzon, J. Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 2006, 57, 303–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Celenza, J.L.; Quiel, J.A.; Smolen, G.A.; Merrikh, H.; Silvestro, A.R.; Normanly, J.; Bender, J. The Arabidopsis ATR1 Myb transcription factor controls indolic glucosinolate homeostasis. Plant Physiol. 2005, 137, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Gigolashvili, T.; Yatusevich, R.; Berger, B.; Muller, C.; Flugge, U.I. The R2R3-MYB transcription factor HAG1/MYB28 is a regulator of methionine-derived glucosinolate biosynthesis in Arabidopsis thaliana. Plant J. 2007, 51, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Gigolashvili, T.; Berger, B.; Mock, H.P.; Muller, C.; Weisshaar, B.; Flugge, U.I. The transcription factor HIG1/MYB51 regulates indolic glucosinolate biosynthesis in Arabidopsis thaliana. Plant J. 2007, 50, 886–901. [Google Scholar] [CrossRef] [Green Version]
- Gigolashvili, T.; Engqvist, M.; Yatusevich, R.; Muller, C.; Flugge, U.I. HAG2/MYB76 and HAG3/MYB29 exert a specific and coordinated control on the regulation of aliphatic glucosinolate biosynthesis in Arabidopsis thaliana. New Phytol. 2008, 177, 627–642. [Google Scholar] [CrossRef]
- Frerigmann, H.; Gigolashvili, T. MYB34, MYB51, and MYB122 distinctly regulate indolic glucosinolate biosynthesis in Arabidopsis thaliana. Mol. Plant 2014, 7, 814–828. [Google Scholar] [CrossRef] [Green Version]
- Velasco, P.; Soengas, P.; Vilar, M.; Cartea, M.E.; del Rio, M. Comparison of Glucosinolate Profiles in Leaf and Seed Tissues of Different Brassica napus Crops. J. Am. Soc. Hortic. Sci. 2008, 133, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Harper, A.L.; Trick, M.; Higgins, J.; Fraser, F.; Clissold, L.; Wells, R.; Hattori, C.; Werner, P.; Bancroft, I. Associative transcriptomics of traits in the polyploid crop species Brassica Napus. Nat. Biotechnol. 2012, 30, 798–802. [Google Scholar] [CrossRef]
- Howell, P.M.; Sharpe, A.G.; Lydiate, D.J. Homoeologous loci control the accumulation of seed glucosinolates in oilseed rape (Brassica napus). Genome 2003, 46, 454–460. [Google Scholar] [CrossRef]
- Li, F.; Chen, B.; Xu, K.; Wu, J.; Song, W.; Bancroft, I.; Harper, A.L.; Trick, M.; Liu, S.; Gao, G.; et al. Genome-Wide Association Study Dissects the Genetic Architecture of Seed Weight and Seed Quality in Rapeseed (Brassica napus L.). DNA Res. 2014, 21, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Harper, A.L.; Trick, M.; Morgan, C.; Fraser, F.; O’Neill, C.; Bancroft, I. Associative Transcriptomics Study Dissects the Genetic Architecture of Seed Glucosinolate Content in Brassica napus. DNA Res. 2014, 21, 613–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, C.M.; Li, S.M.; Duan, X.J.; Fan, J.H.; Jia, L.D.; Zhao, H.Y.; Lu, K.; Li, J.N.; Xu, X.F.; Wang, R. Identification of Candidate Genes for Seed Glucosinolate Content Using Association Mapping in Brassica napus L. Genes 2015, 6, 1215–1229. [Google Scholar] [CrossRef] [Green Version]
- Eskandari, M.; Cober, E.R.; Rajcan, I. Genetic control of soybean seed oil: II. QTL and genes that increase oil concentration without decreasing protein or with increased seed yield. Theor. Appl. Genet. 2013, 126, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Wang, H.; Wang, X.; Guo, L.; Gu, J.; Zhao, W.; Li, B.; Chen, D.; Raboanatahiry, N.; Li, M. Genetic dissection of seed oil and protein content and identification of networks associated with oil content in Brassica napus. Sci. Rep. 2017, 7, 46295. [Google Scholar] [CrossRef] [Green Version]
- Qu, C.; Jia, L.; Fu, F.; Zhao, H.; Lu, K.; Wei, L.; Xu, X.; Liang, Y.; Li, S.; Wang, R.; et al. Genome-wide association mapping and Identification of candidate genes for fatty acid composition in Brassica napus L. using SNP markers. BMC Genom. 2017, 18, 232. [Google Scholar] [CrossRef] [Green Version]
- Ecke, W.; Uzunova, M.; Weissleder, K. Mapping the genome of rapeseed (Brassica napus L.). II. Localization of genes controlling erucic acid synthesis and seed oil content. Theor. Appl. Genet. 1995, 91, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.J.; Barnes, S.R.; Bowman, J.G.; Clarke, M.H.; Werner, C.P.; Kearsey, M.J. QTL analysis of an intervarietal set of substitution lines in Brassica napus: (i) Seed oil content and fatty acid composition. Heredity 2003, 90, 39–48. [Google Scholar] [CrossRef]
- Delourme, R.; Falentin, C.; Huteau, V.; Clouet, V.; Horvais, R.; Gandon, B.; Specel, S.; Hanneton, L.; Dheu, J.E.; Deschamps, M.; et al. Genetic control of oil content in oilseed rape (Brassica napus L.). Theor. Appl. Genet. 2006, 113, 1331–1345. [Google Scholar] [CrossRef]
- Qiu, D.; Morgan, C.; Shi, J.; Long, Y.; Liu, J.; Li, R.; Zhuang, X.; Wang, Y.; Tan, X.; Dietrich, E.; et al. A comparative linkage map of oilseed rape and its use for QTL analysis of seed oil and erucic acid content. Theor. Appl. Genet. 2006, 114, 67–80. [Google Scholar] [CrossRef]
- Yan, X.Y.; Li, J.N.; Fu, F.Y.; Jin, M.Y.; Chen, L.; Liu, L.Z. Co-location of seed oil content, seed hull content and seed coat color QTL in three different environments in Brassica napus L. Euphytica 2009, 170, 355–364. [Google Scholar] [CrossRef]
- Chen, G.; Geng, J.; Rahman, M.; Liu, X.; Tu, J.; Fu, T.; Li, G.; McVetty, P.B.E.; Tahir, M. Identification of QTL for oil content, seed yield, and flowering time in oilseed rape (Brassica napus). Euphytica 2010, 175, 161–174. [Google Scholar] [CrossRef]
- Sun, M.; Hua, W.; Liu, J.; Huang, S.; Wang, X.; Liu, G.; Wang, H. Design of new genome- and gene-sourced primers and identification of QTL for seed oil content in a specially high-oil Brassica napus cultivar. PLoS ONE 2012, 7, e47037. [Google Scholar] [CrossRef]
- Zhao, J.; Huang, J.; Chen, F.; Xu, F.; Ni, X.; Xu, H.; Wang, Y.; Jiang, C.; Wang, H.; Xu, A.; et al. Molecular mapping of Arabidopsis thaliana lipid-related orthologous genes in Brassica napus. Theor. Appl. Genet. 2012, 124, 407–421. [Google Scholar] [CrossRef]
- Si, P.; Mailer, R.J.; Galwey, N.; Turner, D.W. Influence of genotype and environment on oil and protein concentrations of canola (Brassica napus L.) grown across southern Australia. Aust. J. Agric. Res. 2003, 54, 397–407. [Google Scholar] [CrossRef]
- Li, C.; Li, B.; Qu, C.-M.; Yan, X.-Y.; Fu, F.-Y.; Liu, L.-Z.; Chen, L.; Li, J.-N. Analysis of Difference QTLs for Oil Content Between Two Environments in Brassica napus L. Acta Agron. Sin. 2011, 37, 249–254. [Google Scholar] [CrossRef]
- Zhu, Q.; King, G.J.; Liu, X.; Shan, N.; Borpatragohain, P.; Baten, A.; Wang, P.; Luo, S.; Zhou, Q. Identification of SNP loci and candidate genes related to four important fatty acid composition in Brassica napus using genome wide association study. PLoS ONE 2019, 14, e0221578. [Google Scholar] [CrossRef]
- Schneeberger, K.; Ossowski, S.; Lanz, C.; Juul, T.; Petersen, A.H.; Nielsen, K.L.; Jorgensen, J.E.; Weigel, D.; Andersen, S.U. SHOREmap: Simultaneous mapping and mutation identification by deep sequencing. Nat. Methods 2009, 6, 550–551. [Google Scholar] [CrossRef]
- Pucker, B.; Schilbert, H.M. Genomics and Transcriptomics Advance in Plant Sciences. In Molecular Approaches in Plant Biology and Environmental Challenges; Singh, S.P., Upadhyay, S.K., Pandey, A., Kumar, S., Eds.; Energy, Environment, and Sustainability; Springer: Singapore, 2019; pp. 419–448. [Google Scholar]
- Ries, D.; Holtgräwe, D.; Viehöver, P.; Weisshaar, B. Rapid gene identification in sugar beet using deep sequencing of DNA from phenotypic pools selected from breeding panels. BMC Genom. 2016, 17, 236. [Google Scholar] [CrossRef] [Green Version]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Mitsuoka, C.; Tamiru, M.; Innan, H.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yeh, C.T.; Tang, H.M.; Nettleton, D.; Schnable, P.S. Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascher, M.; Jost, M.; Kuon, J.E.; Himmelbach, A.; Assfalg, A.; Beier, S.; Scholz, U.; Graner, A.; Stein, N. Mapping-by-sequencing accelerates forward genetics in barley. Genome Biol. 2014, 15, R78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Yao, J.; Chu, L.; Yuan, Z.; Li, Y.; Zhang, Y. Genetic mapping of the nulliplex-branch gene (gb_nb1) in cotton using next-generation sequencing. Theor. Appl. Genet. 2015, 128, 539–547. [Google Scholar] [CrossRef]
- Itoh, N.; Segawa, T.; Tamiru, M.; Abe, A.; Sakamoto, S.; Uemura, A.; Oikawa, K.; Kutsuzawa, H.; Koga, H.; Imamura, T.; et al. Next-generation sequencing-based bulked segregant analysis for QTL mapping in the heterozygous species Brassica rapa. Theor. Appl. Genet. 2019, 132, 2913–2925. [Google Scholar] [CrossRef]
- Song, J.M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Rousseau-Gueutin, M.; Belser, C.; Da Silva, C.; Richard, G.; Istace, B.; Cruaud, C.; Falentin, C.; Boideau, F.; Boutte, J.; Delourme, R.; et al. Long-read assembly of the Brassica napus reference genome Darmor-bzh. Gigascience 2020, 9, giaa137. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Chawla, H.S.; Obermeier, C.; Dreyer, F.; Abbadi, A.; Snowdon, R. Chromosome-Scale Assembly of Winter Oilseed Rape Brassica napus. Front. Plant Sci. 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Rosso, M.G.; Li, Y.; Strizhov, N.; Reiss, B.; Dekker, K.; Weisshaar, B. An Arabidopsis thaliana T-DNA mutagenised population (GABI-Kat) for flanking sequence tag based reverse genetics. Plant Mol. Biol. 2003, 53, 247–259. [Google Scholar] [CrossRef]
- Andrews, S.; Fast, Q.C. A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 October 2018).
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v1302. Available online: http://arxiv.org/abs/1303.3997.
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 11, 1110. [Google Scholar] [CrossRef]
- Schilbert, H.M.; Rempel, A.; Pucker, B. Comparison of Read Mapping and Variant Calling Tools for the Analysis of Plant NGS Data. Plants 2020, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Pucker, B.; Brockington, S.F. Genome-wide analyses supported by RNA-Seq reveal non-canonical splice sites in plant genomes. BMC Genom. 2018, 19, 980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Pucker, B.; Holtgräwe, D.; Weisshaar, B. Consideration of non-canonical splice sites improves gene prediction on the Arabidopsis thaliana Niederzenz-1 genome sequence. BMC Res. Notes 2017, 10, 667. [Google Scholar] [CrossRef] [Green Version]
- Pucker, B.; Holtgräwe, D.; Rosleff Sörensen, T.; Stracke, R.; Viehöver, P.; Weisshaar, B. A De Novo Genome Sequence Assembly of the Arabidopsis thaliana Accession Niederzenz-1 Displays Presence/Absence Variation and Strong Synteny. PLoS ONE 2016, 11, e0164321. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnke, N.; Suprianto, E.; Möllers, C. A major QTL on chromosome C05 significantly reduces acid detergent lignin (ADL) content and increases seed oil and protein content in oilseed rape (Brassica napus L.). Theor. Appl. Genet. 2018, 131, 2477–2492. [Google Scholar] [CrossRef] [PubMed]
- Schilbert, H.M.; Schöne, M.; Baier, T.; Busche, M.; Viehöver, P.; Weisshaar, B.; Holtgräwe, D. Characterization of the Brassica napus flavonol synthase gene family reveals bifunctional flavonol synthases. Front. Plant Sci. 2021, 12, 733762. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Pucker, B.; Reiher, F.; Schilbert, H.M. Automatic Identification of Players in the Flavonoid Biosynthesis with Application on the Biomedicinal Plant Croton tiglium. Plants 2020, 9, 1103. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.S.; Kim, J.S. Understanding of MYB Transcription Factors Involved in Glucosinolate Biosynthesis in Brassicaceae. Molecules 2017, 22, 1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Bus, A.; Korber, N.; Snowdon, R.J.; Stich, B. Patterns of molecular variation in a species-wide germplasm set of Brassica napus. Theor. Appl. Genet. 2011, 123, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Wang, X. The Arabidopsis Phospholipase D Family. Characterization of a Calcium-Independent and Phosphatidylcholine-Selective PLDζ1 with Distinct Regulatory Domains. Plant Physiol. 2002, 128, 1057–1068. [Google Scholar] [CrossRef] [Green Version]
- Devaiah, S.P.; Pan, X.; Hong, Y.; Roth, M.; Welti, R.; Wang, X. Enhancing seed quality and viability by suppressing phospholipase D in Arabidopsis. Plant J. 2007, 50, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Tao, F.; Li, W. Lipid profiling demonstrates that suppressing Arabidopsis phospholipase Ddelta retards ABA-promoted leaf senescence by attenuating lipid degradation. PLoS ONE 2013, 8, e65687. [Google Scholar] [CrossRef]
- Chen, J.; Li, W.Q.; Jia, Y.X. The Serine Carboxypeptidase-Like Gene SCPL41 Negatively Regulates Membrane Lipid Metabolism in Arabidopsis thaliana. Plants 2020, 9, 696. [Google Scholar] [CrossRef] [PubMed]
- Nützmann, H.W.; Huang, A.; Osbourn, A. Plant metabolic clusters—From genetics to genomics. New Phytol. 2016, 211, 771–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, A.D.; Mullen, R.T.; Mangroo, D. Arabidopsis At2g40730 encodes a cytoplasmic protein involved in nuclear tRNA export. Botany 2011, 89, 175–190. [Google Scholar] [CrossRef]
- Cho, M.; Lee, Z.W.; Cho, H.T. ATP-binding cassette B4, an auxin-efflux transporter, stably associates with the plasma membrane and shows distinctive intracellular trafficking from that of PIN-FORMED proteins. Plant Physiol. 2012, 159, 642–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarling, E.J.; Edwards, P.A. Dancing with the sterols: Critical roles for ABCG1, ABCA1, miRNAs, and nuclear and cell surface receptors in controlling cellular sterol homeostasis. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2012, 1821, 386–395. [Google Scholar] [CrossRef]
- Ducos, E.; Verges, V.; Duge de Bernonville, T.; Blanc, N.; Giglioli-Guivarc’h, N.; Dutilleul, C. Remarkable Evolutionary Conservation of Antiobesity ADIPOSE/WDTC1 Homologs in Animals and Plants. Genetics 2017, 207, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farazi, T.A.; Waksman, G.; Gordon, J.I. The biology and enzymology of protein N-myristoylation. J. Biol. Chem. 2001, 276, 39501–39504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon-Villalobos, L.I.; Nill, C.; Marrocco, K.; Kretsch, T.; Schwechheimer, C. The evolutionarily conserved Arabidopsis thaliana F-box protein AtFBP7 is required for efficient translation during temperature stress. Gene 2007, 392, 106–116. [Google Scholar] [CrossRef]
- Cui, S.; Fukao, Y.; Mano, S.; Yamada, K.; Hayashi, M.; Nishimura, M. Proteomic analysis reveals that the Rab GTPase RabE1c is involved in the degradation of the peroxisomal protein receptor PEX7 (peroxin 7). J. Biol. Chem. 2013, 288, 6014–6023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Fan, C.; Li, J.; Cai, G.; Yang, Q.; Wu, J.; Yi, X.; Zhang, C.; Zhou, Y. A genome-wide association study reveals novel elite allelic variations in seed oil content of Brassica napus. Theor. Appl. Genet. 2016, 129, 1203–1215. [Google Scholar] [CrossRef]
- Nintemann, S.J.; Hunziker, P.; Andersen, T.G.; Schulz, A.; Burow, M.; Halkier, B.A. Localization of the glucosinolate biosynthetic enzymes reveals distinct spatial patterns for the biosynthesis of indole and aliphatic glucosinolates. Physiol. Plant. 2018, 163, 138–154. [Google Scholar] [CrossRef]
- Baskar, V.; Park, S.W. Molecular characterization of BrMYB28 and BrMYB29 paralogous transcription factors involved in the regulation of aliphatic glucosinolate profiles in Brassica rapa ssp. pekinensis. Comptes Rendus Biol. 2015, 338, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-T.; Wu, Z.; Lu, K.; Bi, C.; Liang, S.; Wang, X.-F.; Zhang, D.-P. Overexpression of the MYB transcription factor MYB28 or MYB99 confers hypersensitivity to abscisic acid in Arabidopsis. J. Plant Biol. 2016, 59, 152–161. [Google Scholar] [CrossRef]
- Sarazin, V.; Duclercq, J.; Mendou, B.; Aubanelle, L.; Nicolas, V.; Aono, M.; Pilard, S.; Guerineau, F.; Sangwan-Norreel, B.; Sangwan, R.S. Arabidopsis BNT1, an atypical TIR-NBS-LRR gene, acting as a regulator of the hormonal response to stress. Plant Sci. 2015, 239, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.I.; Park, H.J.; Park, J.H.; Kim, S.; Im, M.Y.; Seo, H.H.; Kim, Y.W.; Hwang, I.; Kim, S.Y. Arabidopsis calcium-dependent protein kinase AtCPK32 interacts with ABF4, a transcriptional regulator of abscisic acid-responsive gene expression, and modulates its activity. Plant Physiol. 2005, 139, 1750–1761. [Google Scholar] [CrossRef] [Green Version]
- Nour-Eldin, H.H.; Andersen, T.G.; Burow, M.; Madsen, S.R.; Jorgensen, M.E.; Olsen, C.E.; Dreyer, I.; Hedrich, R.; Geiger, D.; Halkier, B.A. NRT/PTR transporters are essential for translocation of glucosinolate defence compounds to seeds. Nature 2012, 488, 531–534. [Google Scholar] [CrossRef]
- Kittipol, V.; He, Z.; Wang, L.; Doheny-Adams, T.; Langer, S.; Bancroft, I. Genetic architecture of glucosinolate variation in Brassica napus. J. Plant Physiol. 2019, 240, 152988. [Google Scholar] [CrossRef]
- Wang, B.; Wu, Z.; Li, Z.; Zhang, Q.; Hu, J.; Xiao, Y.; Cai, D.; Wu, J.; King, G.J.; Li, H.; et al. Dissection of the genetic architecture of three seed-quality traits and consequences for breeding in Brassica napus. Plant Biotechnol. J. 2018, 16, 1336–1348. [Google Scholar] [CrossRef] [Green Version]
- Stein, A.; Coriton, O.; Rousseau-Gueutin, M.; Samans, B.; Schiessl, S.V.; Obermeier, C.; Parkin, I.A.P.; Chevre, A.M.; Snowdon, R.J. Mapping of homoeologous chromosome exchanges influencing quantitative trait variation in Brassica napus. Plant Biotechnol. J. 2017, 15, 1478–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, G.E.; Robin, A.H.; Yang, K.; Park, J.I.; Kang, J.G.; Yang, T.J.; Nou, I.S. Identification and expression analysis of glucosinolate biosynthetic genes and estimation of glucosinolate contents in edible organs of Brassica oleracea subspecies. Molecules 2015, 20, 13089–13111. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Chen, H.; Cao, B.; Lei, J.; Chen, G. Molecular Characterization of MYB28 Involved in Aliphatic Glucosinolate Biosynthesis in Chinese Kale (Brassica oleracea var. alboglabra Bailey). Front. Plant Sci. 2017, 8, 1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neequaye, M.; Stavnstrup, S.; Harwood, W.; Lawrenson, T.; Hundleby, P.; Irwin, J.; Troncoso-Rey, P.; Saha, S.; Traka, M.H.; Mithen, R.; et al. CRISPR-Cas9-Mediated Gene Editing of MYB28 Genes Impair Glucoraphanin Accumulation of Brassica oleracea in the Field. CRISPR J. 2021, 4, 416–426. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Variants | Gold-Standard SNVs | |
|---|---|---|---|
| P1 | 3,580,759 | 903,253 (File S4) | |
| P2 | 4,905,445 | ||
| SNVs left after filtering for gold standard | Statistically meaningful differential allele-specific read counts (dARCs) | ||
| SPC_A2 High-pool | 5,215,492 | 889,280 (File S6) | 8407 |
| SPC_A2 Low-pool | 4,848,100 | ||
| GSL High-pool | 5,105,239 | 880,842 (File S5) | 20,726 |
| GSL Low-pool | 5,003,187 |
| Interval ID | Chromosome | Size [bp] | Start [bp] | End [bp] |
|---|---|---|---|---|
| A02_GSL_1 | chrA02 | 326,909 | 23,675,288 | 24,002,197 |
| A06_GSL_1 | chrA06 | 90,191 | 16,818,388 | 16,908,579 |
| A09_GSL_1 | chrA09 | 1,007,698 | 28,668 | 1,036,366 |
| A09_GSL_2 | chrA09 | 479,317 | 1,465,924 | 1,945,241 |
| A09_GSL_3 | chrA09 | 73,214 | 2,071,845 | 2,145,059 |
| A09_GSL_4 | chrA09 | 317,624 | 2,303,455 | 2,621,079 |
| A09_GSL_5 | chrA09 | 248,198 | 3,036,680 | 3,284,878 |
| A09_GSL_6 | chrA09 | 395,239 | 3,440,941 | 3,836,180 |
| A09_GSL_7 | chrA09 | 308,180 | 4,056,024 | 4,364,204 |
| A09_GSL_8 | chrA09 | 79,540 | 4,580,338 | 4,659,878 |
| A09_GSL_9 | chrA09 | 270,234 | 6,924,362 | 7,194,596 |
| A09_GSL_10 | chrA09 | 934,210 | 8,018,931 | 8,953,141 |
| A09_GSL_11 | chrA09 | 215,209 | 9,150,682 | 9,365,891 |
| A09_GSL_12 | chrA09 | 173,036 | 9,514,676 | 9,687,712 |
| A09_GSL_13 | chrA09 | 247,692 | 9,859,341 | 10,107,033 |
| A09_GSL_14 | chrA09 | 78,703 | 10,831,700 | 10,910,403 |
| A09_GSL_15 | chrA09 | 117,588 | 10,993,988 | 11,111,576 |
| A09_GSL_16 | chrA09 | 374,266 | 12,597,247 | 12,971,513 |
| A09_GSL_17 | chrA09 | 710,703 | 13,041,084 | 13,751,787 |
| A09_GSL_18 | chrA09 | 378,004 | 16,764,142 | 17,142,146 |
| C02_GSL_1 | chrC02 | 247,718 | 12,063,724 | 12,311,442 |
| C02_GSL_2 | chrC02 | 724,820 | 44,411,662 | 45,136,482 |
| C02_GSL_3 | chrC02 | 431,332 | 45,205,897 | 45,637,229 |
| C07_GSL_1 | chrC07 | 562,637 | 32,063,223 | 32,625,860 |
| C07_GSL_2 | chrC07 | 204,488 | 35,236,183 | 35,440,671 |
| C09_GSL_1 | chrC09 | 671,097 | 1,152,540 | 1,823,637 |
| C09_GSL_2 | chrC09 | 1,320,909 | 1,911,629 | 3,232,538 |
| C09_GSL_3 | chrC09 | 195,766 | 3,410,309 | 3,606,075 |
| C09_GSL_4 | chrC09 | 425,478 | 3,699,696 | 4,125,174 |
| C09_GSL_5 | chrC09 | 199,414 | 4,285,280 | 4,484,694 |
| Lorenz (P1) | Janetzkis Schlesischer (P2) | |
|---|---|---|
| BnaMYB28 | 4 | 5 |
| BnaMYB29 | 4 | 4 |
| BnaMYB34 | 8 | 7 |
| BnaMYB51 | 7 | 7 |
| BnaMYB122 | 6 | 5 |
| Name | Zheyou7 | Darmor-bzh | Janetzkis 123456Schlesischer (P2) | Lorenz (P1) |
|---|---|---|---|---|
| BnaMYB28_1 | BnaC07T0355800ZY | BnaCnng43220D | Present in genomic mapping | Present in genomic mapping |
| BnaMYB28_2 | BnaC09T0054800ZY | BnaC09g05300D + BnaC09g05290D | Present in genomic mapping | Present in genomic mapping |
| BnaMYB28_3 | BnaA03T0422000ZY | BnaA03g40190D | Present in genomic mapping | Present in genomic mapping |
| BnaMYB28_4 | BnaA09T0074900ZY | Deleted | Absent | Absent |
| BnaMYB28_5 | BnaC02T0362400ZY | Deleted | Present in genomic mapping | Absent |
| BnaMYB28_6 | BnaA02T0409000ZY | Non-functional copy | Present in genomic mapping | Present in genomic mapping |
| Interval ID | Chromosome | Size [bp] | Start [bp] | End [bp] |
|---|---|---|---|---|
| A01_SPC_1 | chrA01 | 143,410 | 15,427,093 | 15,570,503 |
| A06_SPC_1 | chrA06 | 247,164 | 8,434,596 | 8,681,760 |
| A06_SPC_2 | chrA06 | 72,097 | 9,931,989 | 10,004,086 |
| A06_SPC_3 | chrA06 | 410,672 | 10,177,027 | 10,587,699 |
| A09_SPC_1 | chrA09 | 41,585 | 33,415,524 | 33,457,109 |
| C03_SPC_1 | chrC03 | 86,028 | 2,142,025 | 2,228,053 |
| C04_SPC_1 | chrC04 | 53,519 | 364,385 | 417,904 |
| C04_SPC_2 | chrC04 | 10,677 | 1,104,377 | 1,115,054 |
| C08_SPC_1 | chrC08 | 252,499 | 1,743,278 | 1,995,777 |
| C08_SPC_2 | chrC08 | 2,072,658 | 6,332,262 | 8,404,920 |
| C08_SPC_3 | chrC08 | 472,053 | 9,033,205 | 9,505,258 |
| C08_SPC_4 | chrC08 | 464,034 | 12,719,350 | 13,183,384 |
| C08_SPC_5 | chrC08 | 138,554 | 13,409,507 | 13,548,061 |
| C09_SPC_1 | chrC09 | 92,091 | 16,262,887 | 16,354,978 |
| C09_SPC_2 | chrC09 | 381,823 | 17,275,714 | 17,657,537 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schilbert, H.M.; Pucker, B.; Ries, D.; Viehöver, P.; Micic, Z.; Dreyer, F.; Beckmann, K.; Wittkop, B.; Weisshaar, B.; Holtgräwe, D. Mapping-by-Sequencing Reveals Genomic Regions Associated with Seed Quality Parameters in Brassica napus. Genes 2022, 13, 1131. https://doi.org/10.3390/genes13071131
Schilbert HM, Pucker B, Ries D, Viehöver P, Micic Z, Dreyer F, Beckmann K, Wittkop B, Weisshaar B, Holtgräwe D. Mapping-by-Sequencing Reveals Genomic Regions Associated with Seed Quality Parameters in Brassica napus. Genes. 2022; 13(7):1131. https://doi.org/10.3390/genes13071131
Chicago/Turabian StyleSchilbert, Hanna Marie, Boas Pucker, David Ries, Prisca Viehöver, Zeljko Micic, Felix Dreyer, Katrin Beckmann, Benjamin Wittkop, Bernd Weisshaar, and Daniela Holtgräwe. 2022. "Mapping-by-Sequencing Reveals Genomic Regions Associated with Seed Quality Parameters in Brassica napus" Genes 13, no. 7: 1131. https://doi.org/10.3390/genes13071131
APA StyleSchilbert, H. M., Pucker, B., Ries, D., Viehöver, P., Micic, Z., Dreyer, F., Beckmann, K., Wittkop, B., Weisshaar, B., & Holtgräwe, D. (2022). Mapping-by-Sequencing Reveals Genomic Regions Associated with Seed Quality Parameters in Brassica napus. Genes, 13(7), 1131. https://doi.org/10.3390/genes13071131