
Unlocking the Potential of the CA2, CA7, and ITM2C Gene Signatures for the Early Detection of Colorectal Cancer: A Comprehensive Analysis of RNA-Seq Data by Utilizing Machine Learning Algorithms

Abstract
:1. Introduction
2. Materials and Methods
2.1. CRC Gene Expression Dataset Collection
2.2. Feature Selection on CRC Gene Expression Datasets
2.3. Cumulative Gene Expression and Correlation Analysis of the Selected Features
2.4. External CRC Dataset Validation and ROC Curve Analysis
2.5. Analysis of Association of Final Set of Gene Expression with Tumor-Infiltrating Immunocytes
2.6. Biological Function, Literature, and Pathway Analysis of the Selected Gene Signatures
3. Results
3.1. Dataset Overview and Its Preprocessing
3.2. Feature Selection and Machine Learning Model Implementation
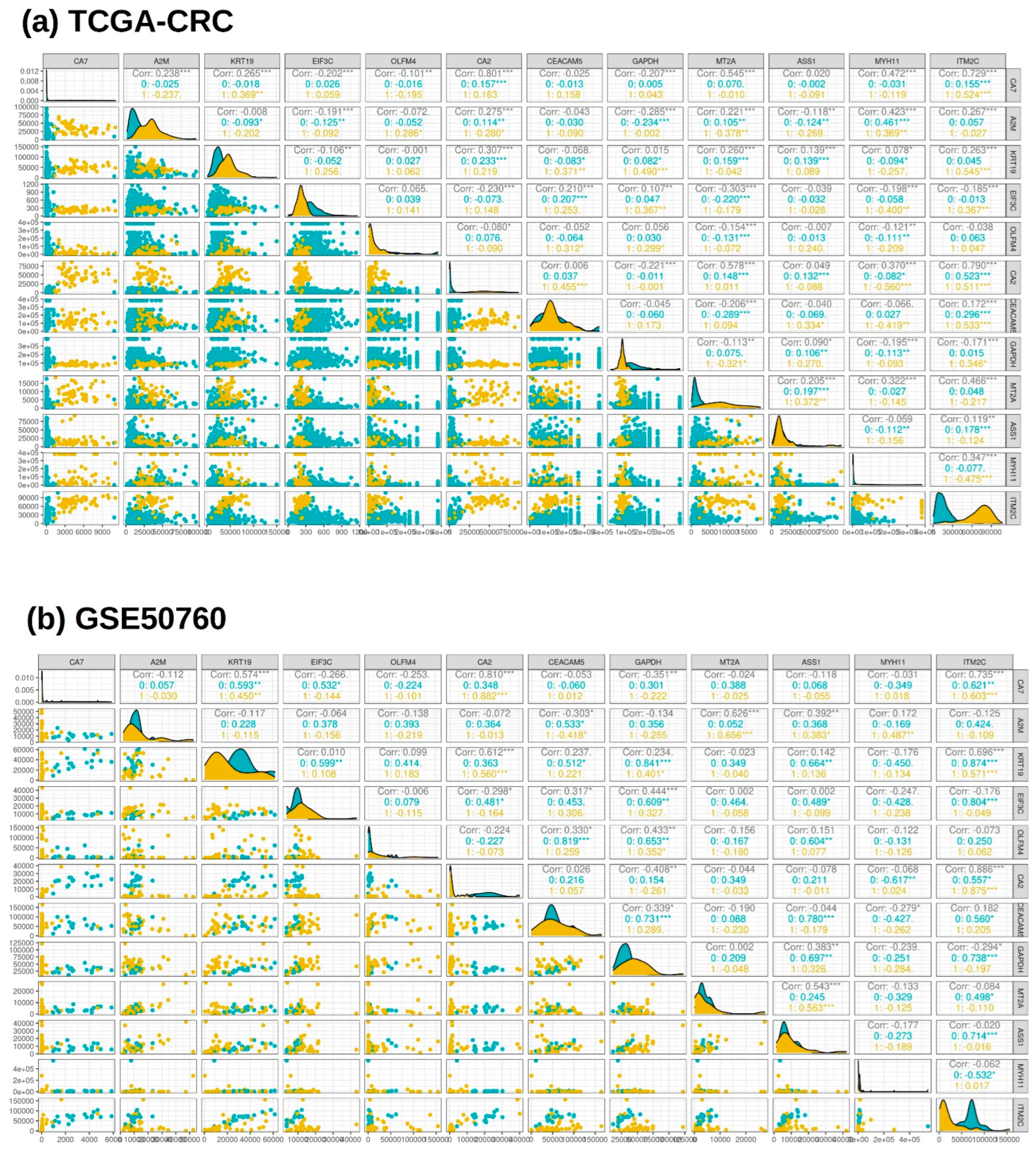
3.3. Cumulative Gene Expression and Correlation Analysis of the Selected Features
3.4. External CRC Dataset Validation and ROC Curve Analysis
3.5. Analysis of Association of Final Set of Gene Expression with Tumor Infiltrating Immunocytes
3.6. Biological Function and Pathway Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cervantes, A.; Adam, R.; Roselló, S.; Arnold, D.; Normanno, N.; Taïeb, J.; Seligmann, J.; De Baere, T.; Osterlund, P.; Yoshino, T. Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 10–32. [Google Scholar] [CrossRef]
- Akimoto, N.; Ugai, T.; Zhong, R.; Hamada, T.; Fujiyoshi, K.; Giannakis, M.; Wu, K.; Cao, Y.; Ng, K.; Ogino, S. Rising incidence of early-onset colorectal cancer—A call to action. Nat. Rev. Clin. Oncol. 2021, 18, 230–243. [Google Scholar] [CrossRef]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Smit, W.L.; Spaan, C.N.; Johannes de Boer, R.; Ramesh, P.; Martins Garcia, T.; Meijer, B.J.; Vermeulen, J.L.M.; Lezzerini, M.; MacInnes, A.W.; Koster, J. Driver mutations of the adenoma-carcinoma sequence govern the intestinal epithelial global translational capacity. Proc. Natl. Acad. Sci. USA 2020, 117, 25560–25570. [Google Scholar] [CrossRef] [PubMed]
- Matas, J.; Kohrn, B.; Fredrickson, J.; Carter, K.; Yu, M.; Wang, T.; Gui, X.; Soussi, T.; Moreno, V.; Grady, W.M. Colorectal cancer is associated with the presence of cancer driver mutations in normal colon. Cancer Res. 2022, 82, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, J.; Wang, X.; Zhao, L.; Wang, Y.; Fan, Z.; Liu, L.; Gao, W. Identification and clinical validation of key genes as the potential biomarkers in colorectal adenoma. BMC Cancer 2023, 23, 39. [Google Scholar] [CrossRef]
- Fu, M.; Huang, Y.; Peng, X.; Li, X.; Luo, N.; Zhu, W.; Yang, F.; Chen, Z.; Ma, S.; Zhang, Y. Development of tumor mutation burden-related prognostic model and novel biomarker identification in stomach adenocarcinoma. Front. Cell Dev. Biol. 2022, 10, 790920. [Google Scholar] [CrossRef]
- Su, Y.; Tian, X.; Gao, R.; Guo, W.; Chen, C.; Chen, C.; Jia, D.; Li, H.; Lv, X. Colon cancer diagnosis and staging classification based on machine learning and bioinformatics analysis. Comput. Biol. Med. 2022, 145, 105409. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Kim, S.-K.; Kim, S.-Y.; Kim, J.-H.; Roh, S.A.; Cho, D.-H.; Kim, Y.S.; Kim, J.C. A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients. Mol. Oncol. 2014, 8, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Ha, Y.J.; Tak, K.H.; Roh, S.A.; Kim, C.W.; Kim, T.W.; Kim, S.-K.; Kim, S.-Y.; Cho, D.-H.; Kim, Y.S. Complex behavior of ALDH1A1 and IGFBP1 in liver metastasis from a colorectal cancer. PLoS ONE 2016, 11, e0155160. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Kim, S.-Y.; Kim, C.W.; Roh, S.A.; Ha, Y.J.; Lee, J.L.; Heo, H.; Cho, D.-H.; Lee, J.-S.; Kim, Y.S. A prognostic index based on an eleven gene signature to predict systemic recurrences in colorectal cancer. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schapire, R.E. Explaining adaboost. In Empirical Inference: Festschrift in Honor of Vladimir N Vapnik; Springer: Berlin/Heidelberg, Germany, 2013; pp. 37–52. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Yan, X.; Su, X. Linear Regression Analysis: Theory and Computing; World Scientific: Singapore, 2009. [Google Scholar]
- Jahromi, A.H.; Taheri, M. A non-parametric mixture of Gaussian naive Bayes classifiers based on local independent features. In Proceedings of the 2017 Artificial Intelligence and Signal Processing Conference (AISP), Shiraz, Iran, 25–27 October 2017; IEEE: Piscataway, NJ, USA, 2017; pp. 209–212. [Google Scholar]
- Cortes, C.; Vapnik, V. Support vector machine. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Vapnik, V. The Nature of Statistical Learning Theory; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1999. [Google Scholar]
- Cristianini, N.; Shawe-Taylor, J. An Introduction to Support Vector Machines and Other Kernel-Based Learning Methods; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Platt, J. Sequential Minimal Optimization: A Fast Algorithm for Training Support Vector Machines; MSR-TR-98-14 1998. Available online: https://www.microsoft.com/en-us/research/publication/sequential-minimal-optimization-a-fast-algorithm-for-training-support-vector-machines/ (accessed on 12 March 2023).
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I. TISIDB: An integrated repository portal for tumor–immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein–protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33 (Suppl. S1), D433–D437. [Google Scholar] [CrossRef]
- Gilmour, K. Perspectives on carbonic anhydrase. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2010, 157, 193–197. [Google Scholar] [CrossRef]
- Cabaleiro-Lago, C.; Lundqvist, M. The effect of nanoparticles on the structure and enzymatic activity of human carbonic anhydrase I and II. Molecules 2020, 25, 4405. [Google Scholar] [CrossRef]
- Zhou, R.; Huang, W.; Yao, Y.; Wang, Y.; Li, Z.; Shao, B.; Zhong, J.; Tang, M.; Liang, S.; Zhao, X. CA II, a potential biomarker by proteomic analysis, exerts significant inhibitory effect on the growth of colorectal cancer cells. Int. J. Oncol. 2013, 43, 611–621. [Google Scholar] [CrossRef]
- Zhang, H.; Zhuo, C.; Zhou, D.; Zhang, F.; Chen, M.; Xu, S.; Chen, Z. Association between the expression of carbonic anhydrase II and clinicopathological features of hepatocellular carcinoma. Oncol. Lett. 2019, 17, 5721–5728. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Huang, Z.; Liao, Z.; He, C.; Fang, X. Low CA II expression is associated with tumor aggressiveness and poor prognosis in gastric cancer patients. Int. J. Clin. Exp. Pathol. 2014, 7, 6716. [Google Scholar] [PubMed]
- Li, X.-J.; Xie, H.-L.; Lei, S.-J.; Cao, H.-Q.; Meng, T.-Y.; Hu, Y.-L. Reduction of CAII expression in gastric cancer: Correlation with invasion and metastasis. Chin. J. Cancer Res. 2012, 24, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-M.; Yao, C.-T.; Chang, Y.-T.; Chou, H.-L.; Chou, Y.-C.; Chen, K.-H.; Terng, H.-J.; Huang, C.-S.; Lee, C.-C.; Su, S.-L. Gene expression profiling of colorectal tumors and normal mucosa by microarrays meta-analysis using prediction analysis of microarray, artificial neural network, classification, and regression trees. Dis. Markers 2014, 2014, 634123. [Google Scholar] [CrossRef]
- Yang, G.-Z.; Hu, L.; Cai, J.; Chen, H.-Y.; Zhang, Y.; Feng, D.; Qi, C.-Y.; Zhai, Y.-X.; Gong, H.; Fu, H. Prognostic value of carbonic anhydrase VII expression in colorectal carcinoma. BMC Cancer 2015, 15, 209. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, T.; Xia, Y.; Wang, Z.; Li, X.; Chen, W. ITM2A as a tumor suppressor and its correlation with PD-L1 in breast cancer. Front. Oncol. 2021, 10, 581733. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
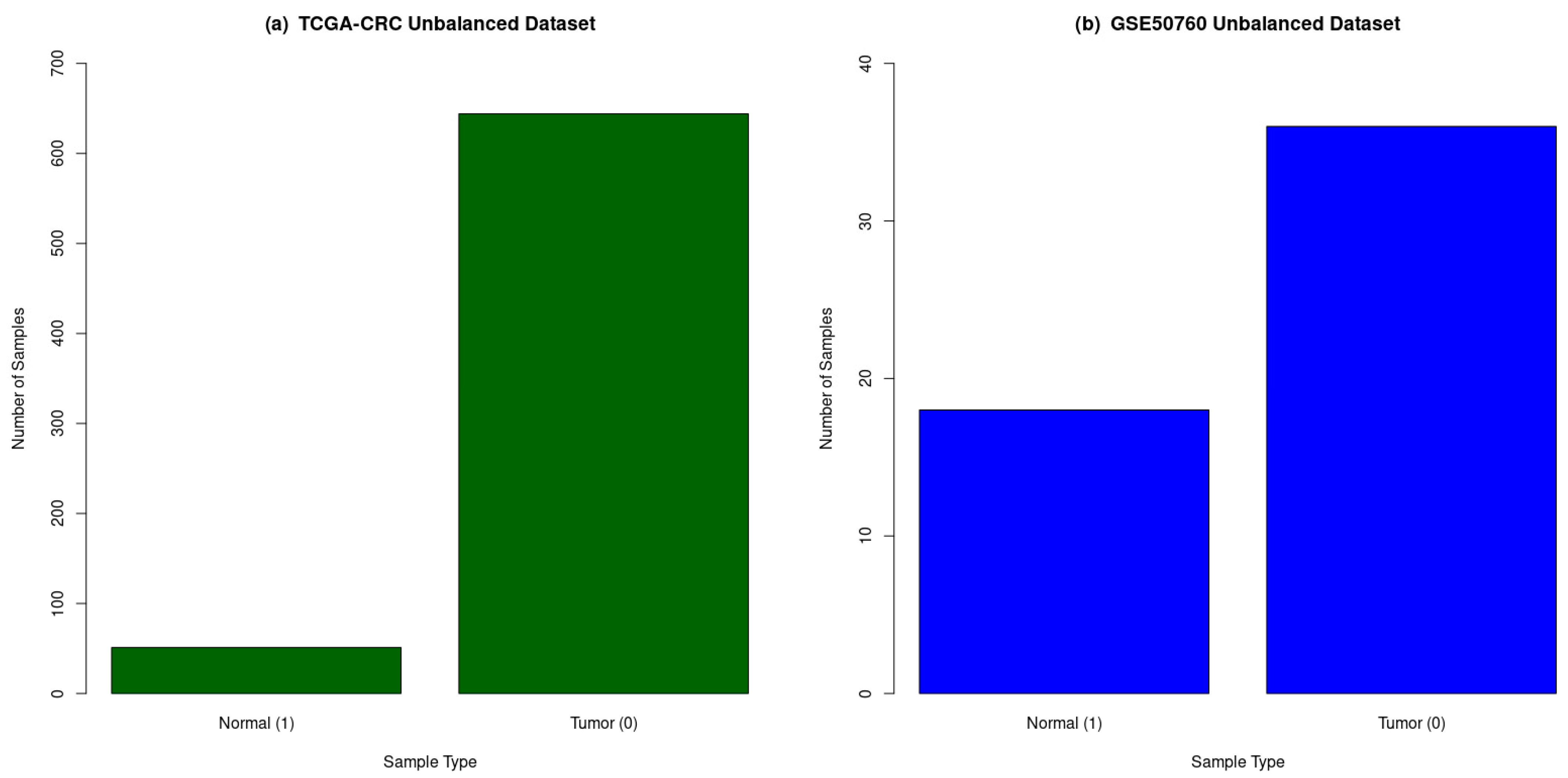
| Sample Groups | Number of Normal Samples | Number of CRC Samples | Total Samples | Number of Genes (Features) |
|---|---|---|---|---|
| TCGA-CRC | 51 | 644 | 695 | 23,187 |
| GSE50760 | 18 | 36 | 54 | 35,238 |
| Sample Groups | ML Algorithms Accuracy with LASSO (%) | ||||
|---|---|---|---|---|---|
| Adaboost | Random Forest | Gaussian Naive Bayes | Support Vector Machine (SVM) | Linear Regression | |
| TCGA-CRC | 99.94 | 100 | 99.93 | 100 | 100 |
| GSE50760 | 94.16 | 94.44 | 93.21 | 93.40 | 92.12 |
| Sample Class | Number of CRC Samples | Precision (PPV) | Recall (Sensitivity) | f1-Score |
|---|---|---|---|---|
| 0 (Tumor) | 6 | 100% | 100% | 100% |
| 1 (Normal) | 6 | 100% | 100% | 100% |
| Gene Name | Biological Function |
|---|---|
| Carbonic anhydrase 2 (CA2) | Carbonic anhydrases are a large family of zinc metalloenzymes that catalyze the reversible hydration of carbon dioxide |
| Carbonic anhydrase 7 (CA7) | |
| Integral membrane protein 2C (ITM2C) | Enables amyloid β binding activity. Involved in negative regulation of neuron projection development and neuron differentiation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maurya, N.S.; Kushwaha, S.; Vetukuri, R.R.; Mani, A. Unlocking the Potential of the CA2, CA7, and ITM2C Gene Signatures for the Early Detection of Colorectal Cancer: A Comprehensive Analysis of RNA-Seq Data by Utilizing Machine Learning Algorithms. Genes 2023, 14, 1836. https://doi.org/10.3390/genes14101836
Maurya NS, Kushwaha S, Vetukuri RR, Mani A. Unlocking the Potential of the CA2, CA7, and ITM2C Gene Signatures for the Early Detection of Colorectal Cancer: A Comprehensive Analysis of RNA-Seq Data by Utilizing Machine Learning Algorithms. Genes. 2023; 14(10):1836. https://doi.org/10.3390/genes14101836
Chicago/Turabian StyleMaurya, Neha Shree, Sandeep Kushwaha, Ramesh Raju Vetukuri, and Ashutosh Mani. 2023. "Unlocking the Potential of the CA2, CA7, and ITM2C Gene Signatures for the Early Detection of Colorectal Cancer: A Comprehensive Analysis of RNA-Seq Data by Utilizing Machine Learning Algorithms" Genes 14, no. 10: 1836. https://doi.org/10.3390/genes14101836
APA StyleMaurya, N. S., Kushwaha, S., Vetukuri, R. R., & Mani, A. (2023). Unlocking the Potential of the CA2, CA7, and ITM2C Gene Signatures for the Early Detection of Colorectal Cancer: A Comprehensive Analysis of RNA-Seq Data by Utilizing Machine Learning Algorithms. Genes, 14(10), 1836. https://doi.org/10.3390/genes14101836