
Independent COL17A1 Variants in Cats with Junctional Epidermolysis Bullosa
 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
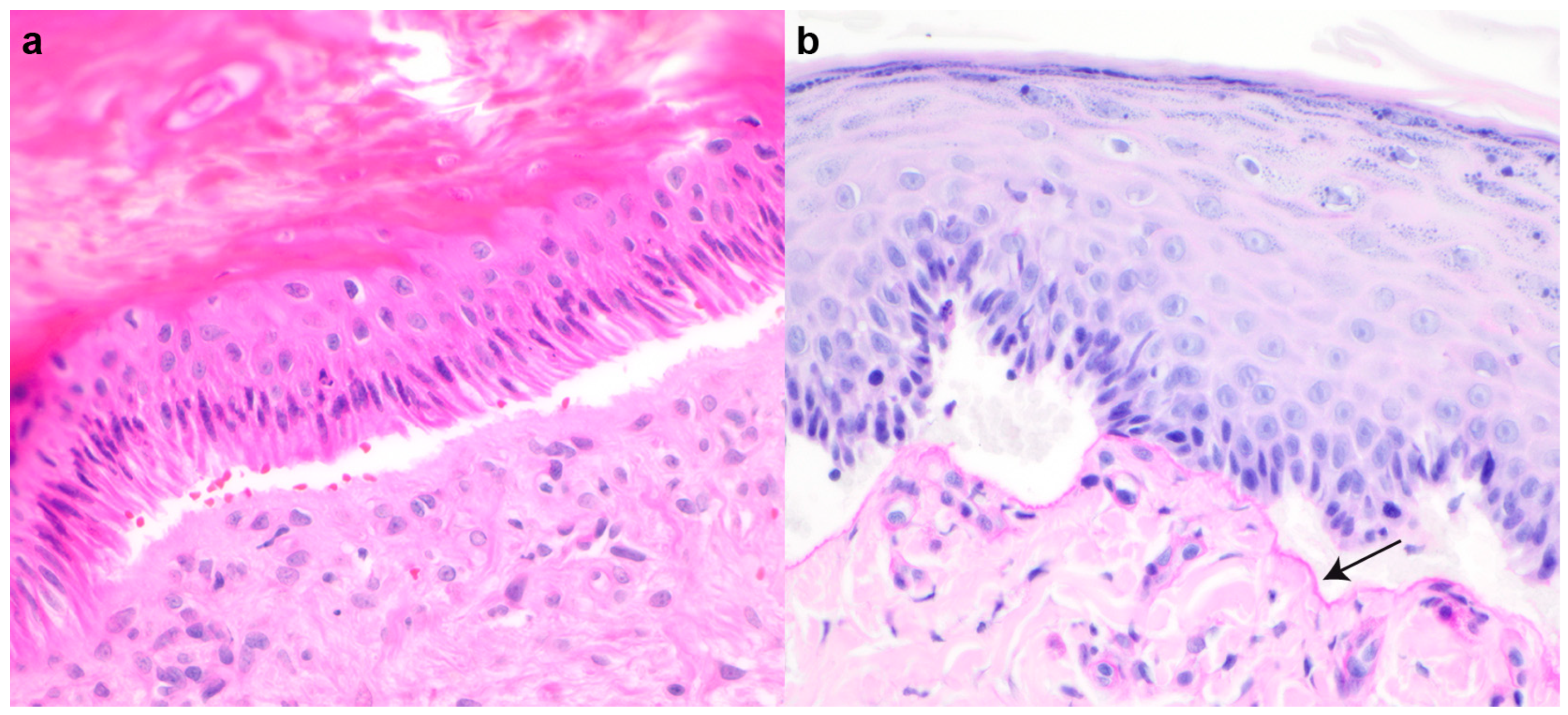
2.2. Histopathological Examinations
2.3. Animals for Genetic Analyses
2.4. DNA Isolation and Whole Genome Sequencing
2.5. Variant Filtering
2.6. Targeted Genotyping
2.7. RNA Extraction and RT-PCR
2.8. Bioinformatic Prediction of Functional Effects on Splicing
3. Results
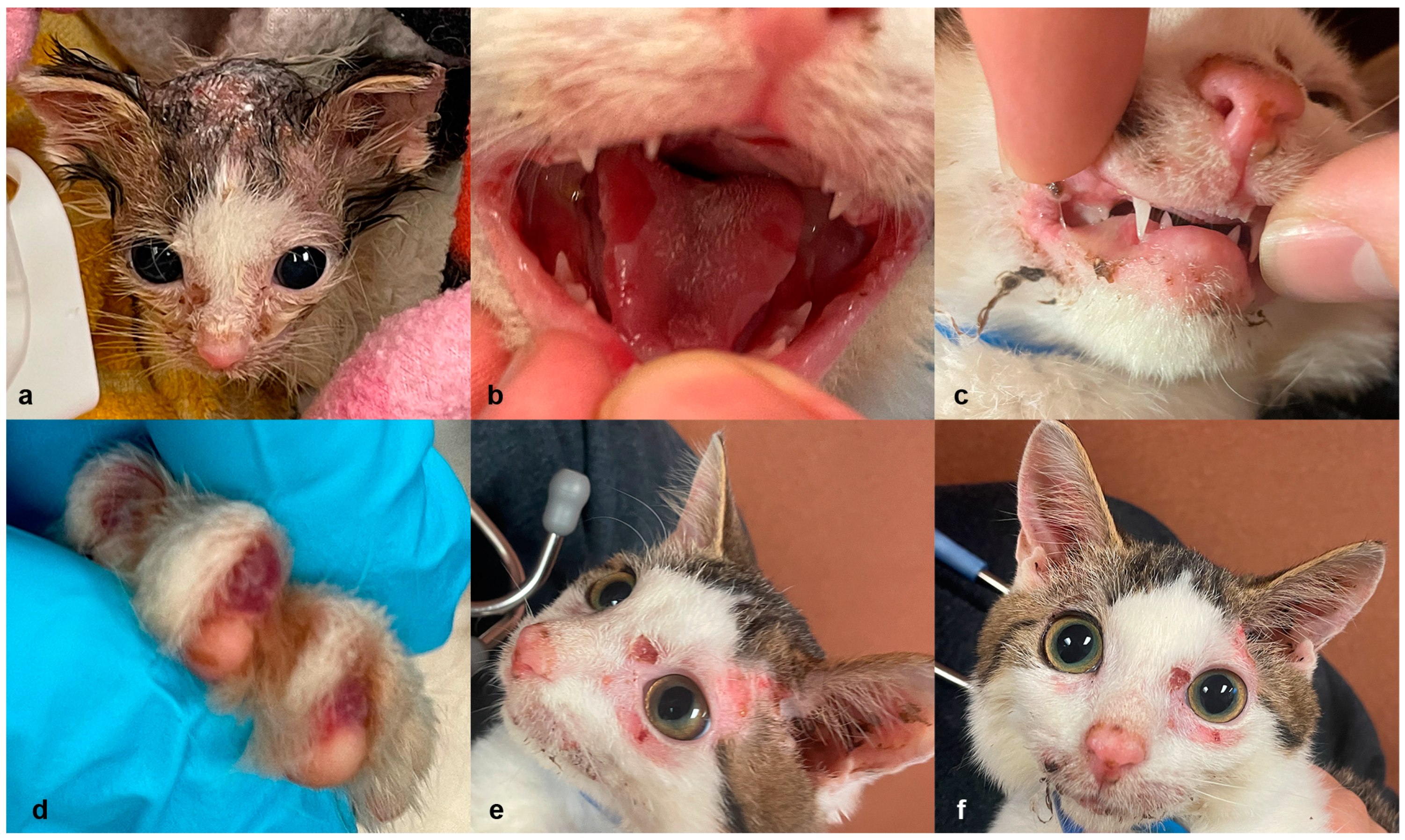
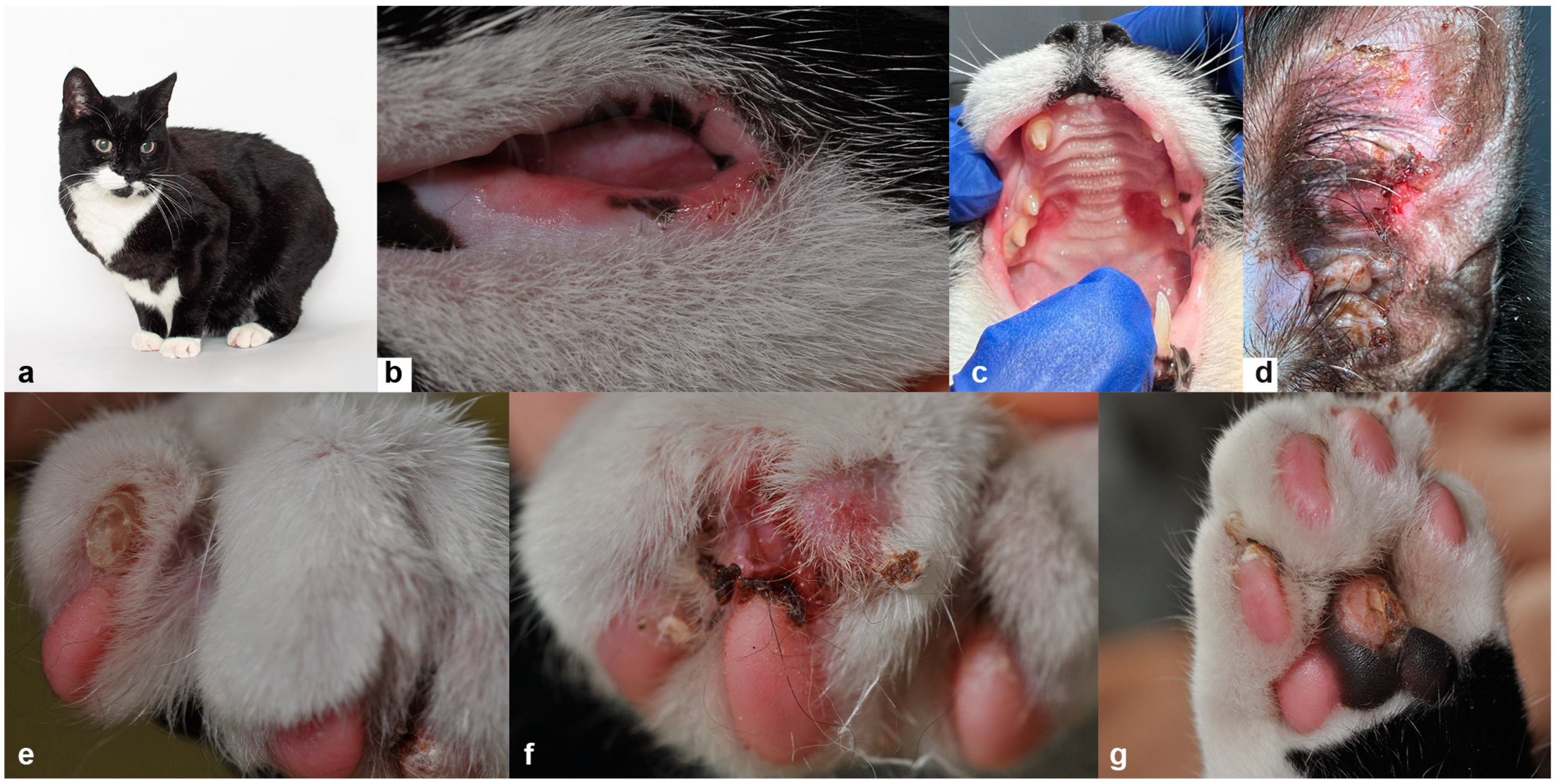
3.1. Clinical and Histopathological Phenotype Characterization
3.2. Genetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Has, C.; Fischer, J. Inherited Epidermolysis Bullosa: New Diagnostics and New Clinical Phenotypes. Exp. Dermatol. 2019, 28, 1146–1152. [Google Scholar] [CrossRef]
- Medeiros, G.X.; Riet-Correa, F. Epidermolysis Bullosa in Animals: A Review. Vet. Dermatol. 2015, 26, 3-e2. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Hess, M.; Anemüller, W.; Blume-Peytavi, U.; Emmert, S.; Fölster-Holst, R.; Frank, J.; Giehl, K.; Günther, C.; Hammersen, J.; et al. Epidemiology of Inherited Epidermolysis Bullosa in Germany. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 402–410. [Google Scholar] [CrossRef]
- Koebner, H. Hereditaeare Anlage Zur Blasenbildung (Epidermolysis Bullosa Hereditaria). Dtsch. Med. Wochenschr. 1886, 12, 21–22. [Google Scholar] [CrossRef]
- Alley, M.R.; O’Hara, P.J.; Middelberg, A. An Epidermolysis Bullosa of Sheep. N. Zeal. Vet. J. 1974, 22, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W.; Schultz, R.D. Epidermolysis Bullosa Simplex in the Collie Dog. J. Am. Vet. Med. Assoc. 1977, 171, 721–727. [Google Scholar]
- Frame, S.R.; Harrington, D.D.; Fessler, J.; Frame, P.F. Hereditary Junctional Mechanobullous Disease in a Foal. J. Am. Vet. Med. Assoc. 1988, 193, 1420–1424. [Google Scholar] [PubMed]
- Thompson, K.G.; Crandell, R.A.; Rugeley, W.W.; Sutherland, R.J. A Mechanobullous Disease with Sub-Basilar Separation in Brangus Calves. Vet. Pathol. 1985, 22, 283–285. [Google Scholar] [CrossRef] [PubMed]
- O’Dair, H.A.; Henderson, J.P. Suspected Mechanobullous Skin Disease in a Cat. J. Small Anim. Pract. 1994, 35, 24–27. [Google Scholar] [CrossRef]
- White, S.D.; Dunstan, R.W.; Olivry, T.; Naydan, D.K.; Richter, K. Dystrophic (Dermolytic) Epidermolysis Bullosa in a Cat. Vet. Dermatol. 1993, 4, 91–95. [Google Scholar] [CrossRef]
- Hammersen, J.; Has, C.; Naumann-Bartsch, N.; Stachel, D.; Kiritsi, D.; Söder, S.; Tardieu, M.; Metzler, M.; Bruckner-Tuderman, L.; Schneider, H. Genotype, Clinical Course, and Therapeutic Decision Making in 76 Infants with Severe Generalized Junctional Epidermolysis Bullosa. J. Investig. Dermatol. 2016, 136, 2150–2157. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.-D. Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatol. 2016, 152, 1231. [Google Scholar] [CrossRef] [PubMed]
- Alhaidari, Z.; Olivry, T.; Spadafora, A.; Thomas, R.C.; Perrin, C.; Meneguzzi, G.; Ortonne, J.P. Junctional Epidermolysis Bullosa in Two Domestic Shorthair Kittens. Vet. Dermatol. 2005, 16, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Doval, I.; Davila-Seijo, P.; Langan, S.M. Updated Systematic Review of Randomized Controlled Trials of Treatments for Inherited Forms of Epidermolysis Bullosa. Clin. Exp. Dermatol. 2013, 38, 92–94. [Google Scholar] [CrossRef]
- Mauldin, E.A.; Wang, P.; Olivry, T.; Henthorn, P.S.; Casal, M.L. Epidermolysis Bullosa Simplex in Sibling Eurasier Dogs Is Caused by a PLEC Non-Sense Variant. Vet. Dermatol. 2017, 28, 10-e3. [Google Scholar] [CrossRef]
- Ford, C.A.; Stanfield, A.M.; Spelman, R.J.; Smits, B.; Ankersmidt-Udy, A.E.L.; Cottier, K.; Holloway, H.; Walden, A.; Al-Wahb, M.; Bohm, E.; et al. A Mutation in Bovine Keratin 5 Causing Epidermolysis Bullosa Simplex, Transmitted by a Mosaic Sire. J. Investig. Dermatol. 2005, 124, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, J.G.P.; Häfliger, I.M.; Veiga, I.M.B.; Drögemüller, C.; Agerholm, J.S. A de Novo Mutation in KRT5 in a Crossbred Calf with Epidermolysis Bullosa Simplex. J. Vet. Intern. Med. 2020, 34, 2800–2807. [Google Scholar] [CrossRef] [PubMed]
- Kiener, S.; Mauldin, E.A.; Jagannathan, V.; Casal, M.L.; Leeb, T. KRT5 Missense Variant in a Cardigan Welsh Corgi with Epidermolysis Bullosa Simplex. Anim. Genet. 2022, 53, 892–896. [Google Scholar] [CrossRef]
- Dettwiler, M.; Leuthard, F.; Bauer, A.; Jagannathan, V.; Lourenço, A.M.; Pereira, H.; Leeb, T.; Welle, M.M. A Nonsense Variant in the KRT14 Gene in a Domestic Shorthair Cat with Epidermolysis Bullosa Simplex. Anim. Genet. 2020, 51, 829–832. [Google Scholar] [CrossRef]
- Menoud, A.; Welle, M.; Tetens, J.; Lichtner, P.; Drögemüller, C. A COL7A1 Mutation Causes Dystrophic Epidermolysis Bullosa in Rotes Höhenvieh Cattle. PLoS ONE 2012, 7, e38823. [Google Scholar] [CrossRef]
- Baldeschi, C.; Gache, Y.; Rattenholl, A.; Bouille, P.; Danos, O.; Ortonne, J.-P.; Bruckner-Tuderman, L.; Meneguzzi, G. Genetic Correction of Canine Dystrophic Epidermolysis Bullosa Mediated by Retroviral Vectors. Hum. Mol. Genet. 2003, 12, 1897–1905. [Google Scholar] [CrossRef]
- Capt, A.; Spirito, F.; Guaguere, E.; Spadafora, A.; Ortonne, J.-P.; Meneguzzi, G. Inherited Junctional Epidermolysis Bullosa in the German Pointer: Establishment of a Large Animal Model. J. Investig. Dermatol. 2005, 124, 530–535. [Google Scholar] [CrossRef]
- Graves, K.T.; Henney, P.J.; Ennis, R.B. Partial Deletion of the LAMA3 Gene Is Responsible for Hereditary Junctional Epidermolysis Bullosa in the American Saddlebred Horse. Anim. Genet. 2009, 40, 35–41. [Google Scholar] [CrossRef]
- Sartelet, A.; Harland, C.; Tamma, N.; Karim, L.; Bayrou, C.; Li, W.; Ahariz, N.; Coppieters, W.; Georges, M.; Charlier, C. A Stop-Gain in the Laminin, Alpha 3 Gene Causes Recessive Junctional Epidermolysis Bullosa in Belgian Blue Cattle. Anim. Genet. 2015, 46, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Kiener, S.; Laprais, A.; Mauldin, E.A.; Jagannathan, V.; Olivry, T.; Leeb, T. LAMB3 Missense Variant in Australian Shepherd Dogs with Junctional Epidermolysis Bullosa. Genes 2020, 11, 1055. [Google Scholar] [CrossRef] [PubMed]
- Spirito, F.; Charlesworth, A.; Ortonne, J.-P.; Meneguzzi, G.; Linder, K.; Baird, J. Animal Models for Skin Blistering Conditions: Absence of Laminin 5 Causes Hereditary Junctional Mechanobullous Disease in the Belgian Horse. J. Investig. Dermatol. 2002, 119, 684–691. [Google Scholar] [CrossRef]
- Mömke, S.; Kerkmann, A.; Wöhlke, A.; Ostmeier, M.; Hewicker-Trautwein, M.; Ganter, M.; Kijas, J.; Distl, O. A Frameshift Mutation within LAMC2 Is Responsible for Herlitz Type Junctional Epidermolysis Bullosa (HJEB) in Black Headed Mutton Sheep. PLoS ONE 2011, 6, e18943. [Google Scholar] [CrossRef]
- Murgiano, L.; Wiedemar, N.; Jagannathan, V.; Isling, L.K.; Drögemüller, C.; Agerholm, J.S. Epidermolysis Bullosa in Danish Hereford Calves Is Caused by a Deletion in LAMC2 Gene. BMC Vet. Res. 2015, 11, 23. [Google Scholar] [CrossRef]
- McGrath, J.A.; Gatalica, B.; Christiano, A.M.; Si, K.; Owaribe, K.; McMillan, J.R.; Eady, R.A.J.; Uitto, J. Mutations in the 180–KD Bullous Pemphigoid Antigen (BPAG2), a Hemidesmosomal Transmembrane Collagen (COL17A1), in Generalized Atrophic Benign Epidermolysis Bullosa. Nat. Genet. 1995, 11, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Olivry, T.; Poujade-Delverdier, A.; Dunston, S.M.; Fine, J.; Ortonne, J. Absent Expression of Collagen XVII (BPAG2, BP180) in Canine Familial Localized Junctional Epidermolysis Bullosa. Vet. Dermatol. 1997, 8, 203–212. [Google Scholar] [CrossRef]
- Gatalica, B.; Pulkkinen, L.; Li, K.; Kuokkanen, K.; Ryynänen, M.; McGrath, J.A.; Uitto, J. Cloning of the Human Type XVII Collagen Gene (COL17A1), and Detection of Novel Mutations in Generalized Atrophic Benign Epidermolysis Bullosa. Am. J. Hum. Genet. 1997, 60, 352–365. [Google Scholar] [PubMed]
- Natsuga, K.; Watanabe, M.; Nishie, W.; Shimizu, H. Life before and beyond Blistering: The Role of Collagen XVII in Epidermal Physiology. Exp. Dermatol. 2019, 28, 1135–1141. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus Reclassification of Inherited Epidermolysis Bullosa and Other Disorders with Skin Fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef]
- Letko, A.; Strugnell, B.; Häfliger, I.M.; Paris, J.M.; Waine, K.; Drögemüller, C.; Scholes, S. Compound Heterozygous PLA2G6 Loss-of-Function Variants in Swaledale Sheep with Neuroaxonal Dystrophy. Mol. Genet. Genom. 2021, 296, 235–242. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef]
- SpliceAI Web Server at the Broad Institute. Available online: https://spliceailookup.broadinstitute.org/ (accessed on 14 September 2023).
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef]
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional Annotations Improve the Predictive Score of Human Disease-Related Mutations in Proteins. Hum. Mutat. 2009, 30, 1237–1244. [Google Scholar] [CrossRef]
- Niroula, A.; Urolagin, S.; Vihinen, M. PON-P2: Prediction Method for Fast and Reliable Identification of Harmful Variants. PLoS ONE 2015, 10, e0117380. [Google Scholar] [CrossRef]
- Johnstone, I.; Mason, K.; Sutton, R. A Hereditary Junctional Mechanobullous Disease in the Cat. In Proceedings of the Second World Congress of Veterinary Dermatology, Montréal, QC, Canada, 13–16 May 1992; World Congress of Veterinary Dermatology Association: Montréal, QC, Canada, 1992; pp. 111–112. [Google Scholar]
- Lourdes Frehner, B.; Christen, M.; Reichler, I.M.; Jagannathan, V.; Novacco, M.; Riond, B.; Peters, L.M.; Suárez Sánchez-Andrade, J.; Pieńkowska-Schelling, A.; Schelling, C.; et al. Autosomal Recessive Hyposegmentation of Granulocytes in Australian Shepherd Dogs Indicates a Role for LMBR1L in Myeloid Leukocytes. PLoS Genet. 2023, 19, e1010805. [Google Scholar] [CrossRef] [PubMed]
- Rudd Garces, G.; Turba, M.E.; Muracchini, M.; Diana, A.; Jagannathan, V.; Gentilini, F.; Leeb, T. PRKG2 Splice Site Variant in Dogo Argentino Dogs with Disproportionate Dwarfism. Genes 2021, 12, 1489. [Google Scholar] [CrossRef]
- Velardo, D.; Antognozzi, S.; Rimoldi, M.; Pagliarani, S.; Cogiamanian, F.; Barbieri, S.; Corti, S.; Comi, G.P.; Ronchi, D. Case Report: Clinical and Molecular Characterization of Two Siblings Affected by Brody Myopathy. Front. Neurol. 2023, 14, 1170071. [Google Scholar] [CrossRef] [PubMed]
- Jenni, S.; Ludwig-Peisker, O.; Jagannathan, V.; Lapsina, S.; Stirn, M.; Hofmann-Lehmann, R.; Bogdanov, N.; Schetle, N.; Giger, U.; Leeb, T.; et al. Methemoglobinemia, Increased Deformability and Reduced Membrane Stability of Red Blood Cells in a Cat with a CYB5R3 Splice Defect. Cells 2023, 12, 991. [Google Scholar] [CrossRef] [PubMed]
- Roca, X.; Olson, A.J.; Rao, A.R.; Enerly, E.; Kristensen, V.N.; Børresen-Dale, A.-L.; Andresen, B.S.; Krainer, A.R.; Sachidanandam, R. Features of 5′-Splice-Site Efficiency Derived from Disease-Causing Mutations and Comparative Genomics. Genome Res. 2008, 18, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Roca, X.; Krainer, A.R.; Eperon, I.C. Pick One, but Be Quick: 5′ Splice Sites and the Problems of Too Many Choices. Genes Dev. 2013, 27, 129–144. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Filtering Step | Variants Case 1 | Variants Case 2 | ||
|---|---|---|---|---|
| Het | Hom | Het | Hom | |
| All variants | 6,364,334 | 5,790,564 | 6,976,390 | 5,784,208 |
| Private variants (allele frequency of 0 in control cohort) | 61,931 | 13,136 | 88,531 | 16,633 |
| Protein-changing private variants | 369 | 83 | 573 | 90 |
| Protein-changing variants in 16 candidate genes | 0 | 1 | 1 | 1 |
| Cat | HGVS-g 1 | HGVS-c 2 | HGVS-r 3 | HGVS-p 4 |
|---|---|---|---|---|
| NC_058378.1 | XM_006938156.5 | XM_006938156.5 | XP_006938218.3 | |
| Case 1 | ChrD2:62,124,169del | c.3019+1del | r.spl? | n.a. |
| Case 2 | ChrD2:62,149,308C>T | c.769+5G>A | r.[=,r.769_770insguacaug] | p.([=,p.Val257Glyfs*82]) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiener, S.; Troyer, H.; Ruvolo, D.; Grest, P.; Soto, S.; Letko, A.; Jagannathan, V.; Leeb, T.; Mauldin, E.A.; Yang, C.; et al. Independent COL17A1 Variants in Cats with Junctional Epidermolysis Bullosa. Genes 2023, 14, 1835. https://doi.org/10.3390/genes14101835
Kiener S, Troyer H, Ruvolo D, Grest P, Soto S, Letko A, Jagannathan V, Leeb T, Mauldin EA, Yang C, et al. Independent COL17A1 Variants in Cats with Junctional Epidermolysis Bullosa. Genes. 2023; 14(10):1835. https://doi.org/10.3390/genes14101835
Chicago/Turabian StyleKiener, Sarah, Heather Troyer, Daniel Ruvolo, Paula Grest, Sara Soto, Anna Letko, Vidhya Jagannathan, Tosso Leeb, Elizabeth A. Mauldin, Ching Yang, and et al. 2023. "Independent COL17A1 Variants in Cats with Junctional Epidermolysis Bullosa" Genes 14, no. 10: 1835. https://doi.org/10.3390/genes14101835
APA StyleKiener, S., Troyer, H., Ruvolo, D., Grest, P., Soto, S., Letko, A., Jagannathan, V., Leeb, T., Mauldin, E. A., Yang, C., & Rostaher, A. (2023). Independent COL17A1 Variants in Cats with Junctional Epidermolysis Bullosa. Genes, 14(10), 1835. https://doi.org/10.3390/genes14101835