




Genes 2024, 15(11), 1360; https://doi.org/10.3390/genes15111360 - 23 Oct 2024
Viewed by 510
Abstract
Background/Objectives: The giant red shrimp, Aristaeomorpha foliacea, is a valuable marine fishing resource. The conservation of species, especially exploited ones, depends on a good knowledge of their biology, as well as the development of appropriate management plans based on the identification of
[...] Read more.
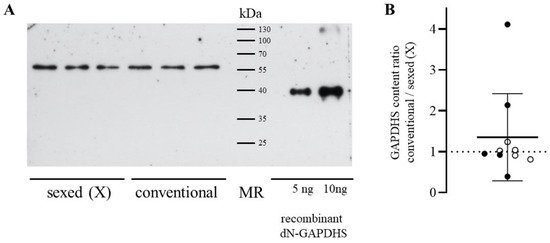
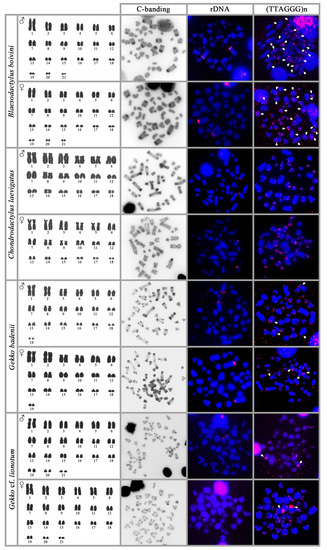
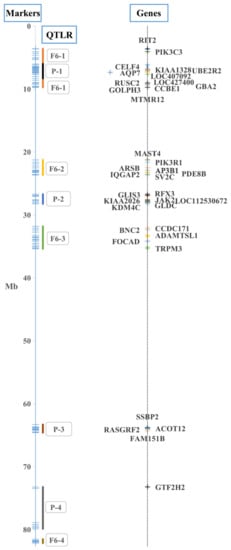
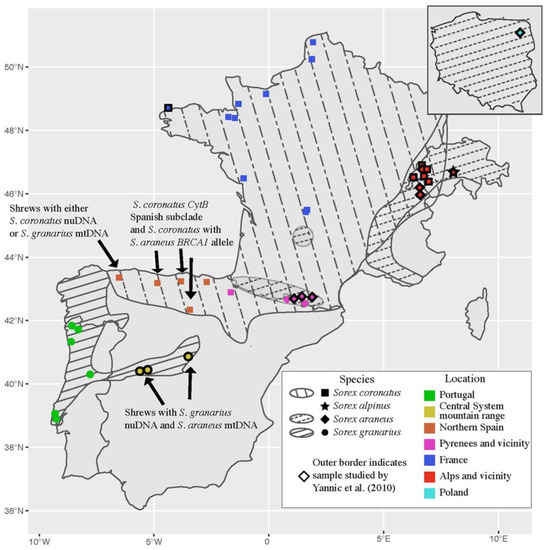
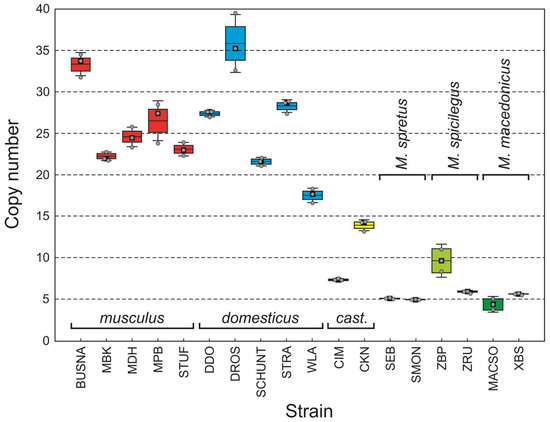
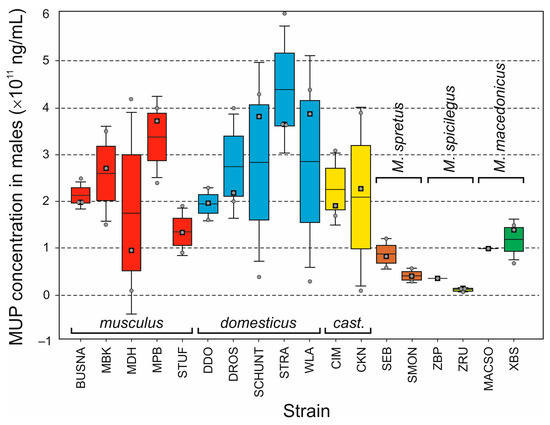
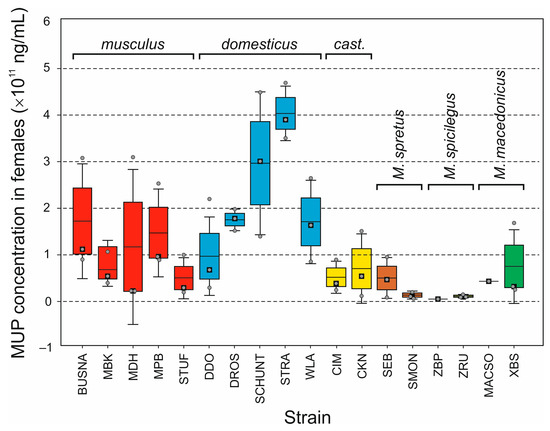
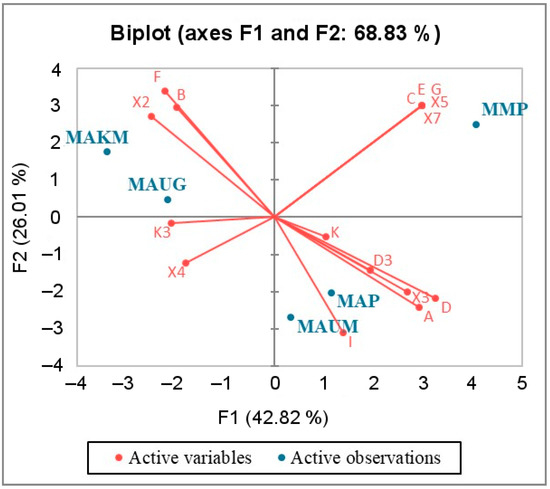
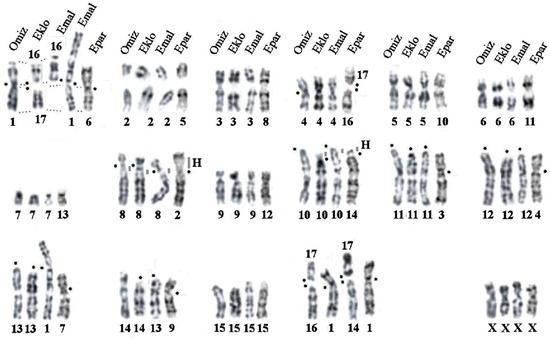
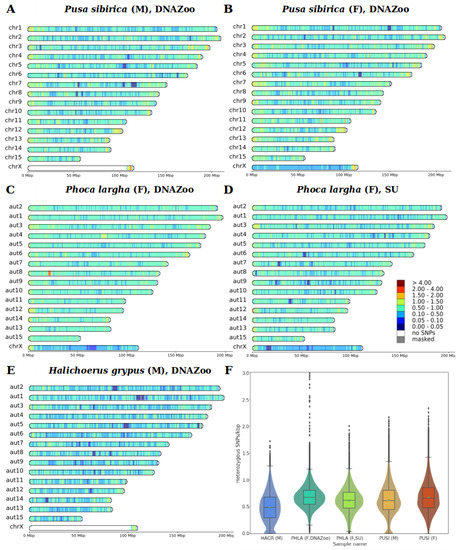
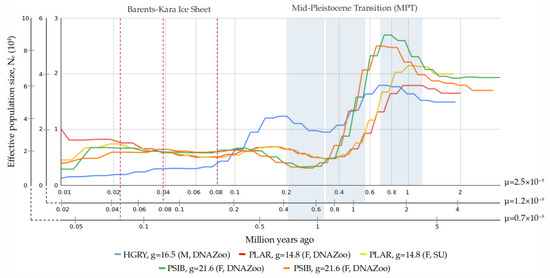
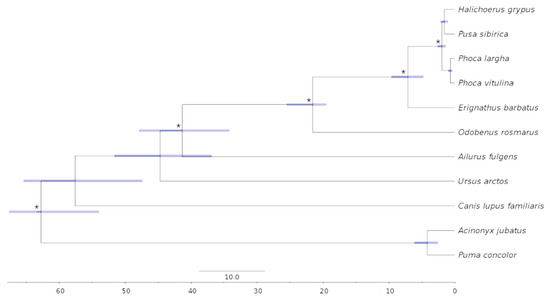
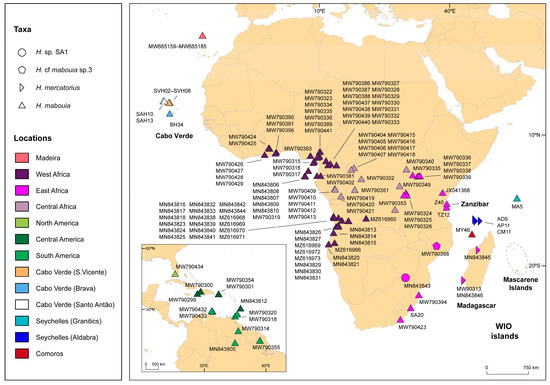
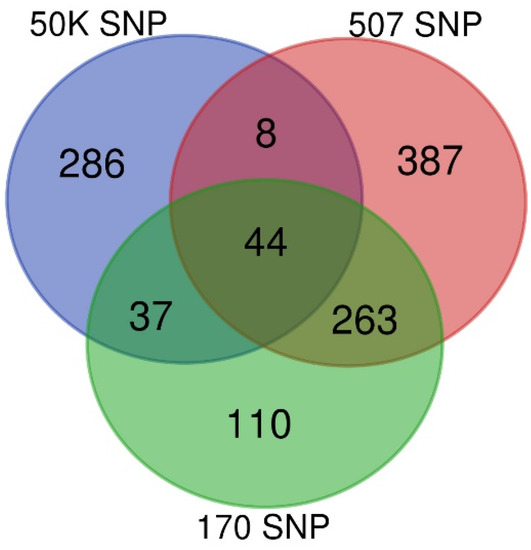
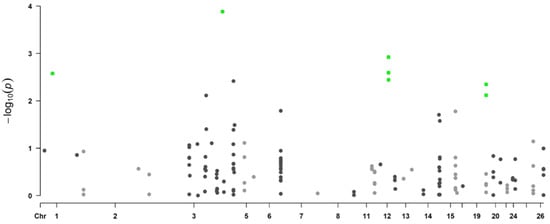
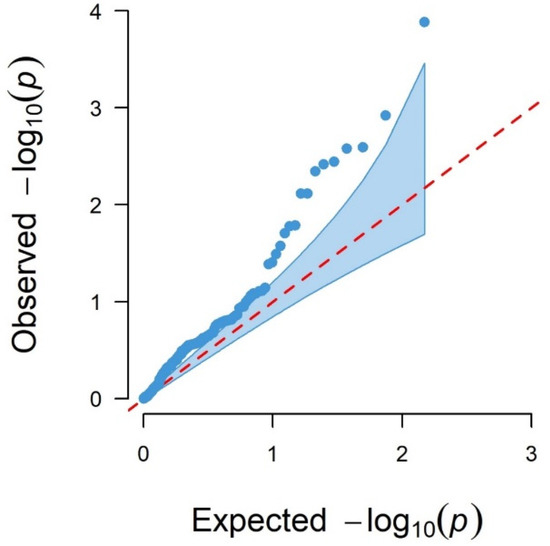
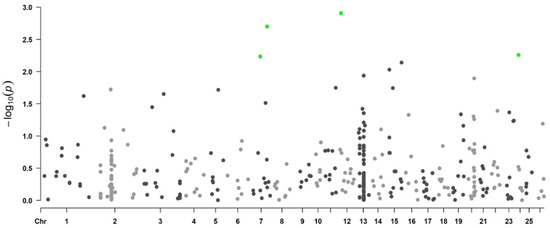
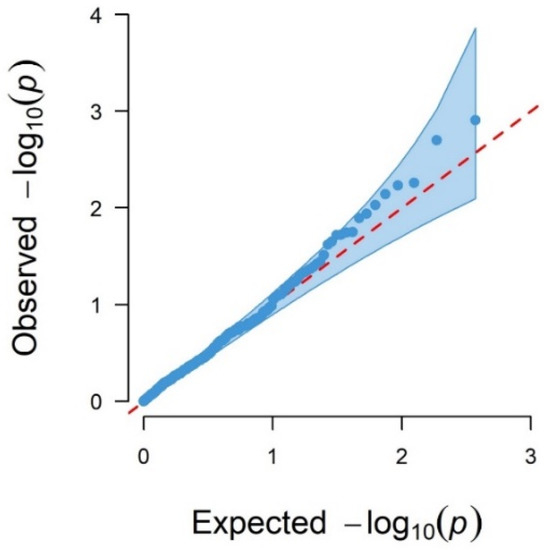
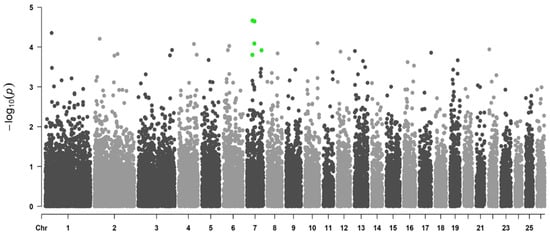
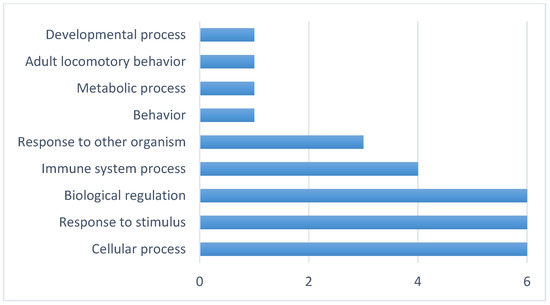
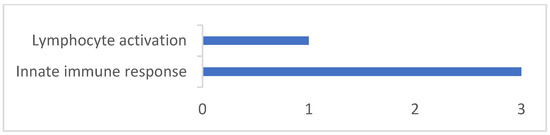
Background/Objectives: The giant red shrimp, Aristaeomorpha foliacea, is a valuable marine fishing resource. The conservation of species, especially exploited ones, depends on a good knowledge of their biology, as well as the development of appropriate management plans based on the identification of genetically differentiated units or genetic stocks. Microsatellites are widely used molecular markers to detect genetic stocks in penaeoid shrimps and prawns. This study aimed to develop and characterize new microsatellites for A. foliacea. Methods: Next-generation sequencing based on 454 pyrosequencing revealed 58 candidate microsatellite loci for A. foliacea. These were tested on a panel of 8 individuals representative of its worldwide geographical distribution, and 19 polymorphic loci were identified and subsequently validated and characterized in 30 individuals from a single population in the Mediterranean Sea. Results: As a result, 10 polymorphic loci were identified, which did not present linkage disequilibrium and showed a range of alleles per locus and an observed and expected heterozygosity of 2–10, 0.0667–0.5567, and 0.0661–0.8511, respectively. Nine out of these loci were under Hardy–Weinberg equilibrium and showed a combined exclusion probability of 0.9202 and 0.9968 in parentage and identity analysis, respectively. Conclusions: This set of loci will provide a strong set of tools to (i) perform parentage studies and (ii) examine connectivity patterns (horizontal and vertical), including examining the population structure of this species at a variety of geographical scales and, particularly, between exploited populations in shallow waters and deeper unexploited populations.
Full article








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}