
Decoding Cancer Evolution: Integrating Genetic and Non-Genetic Insights


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Models of Cancer Evolution: From Linear Succession to Punctuated Equilibrium
3. Cancer Initiation and Progression as a Reverse Microevolutionary Process
4. Diverse Dimensions in Cancer Evolution: Beyond Genetic Drivers
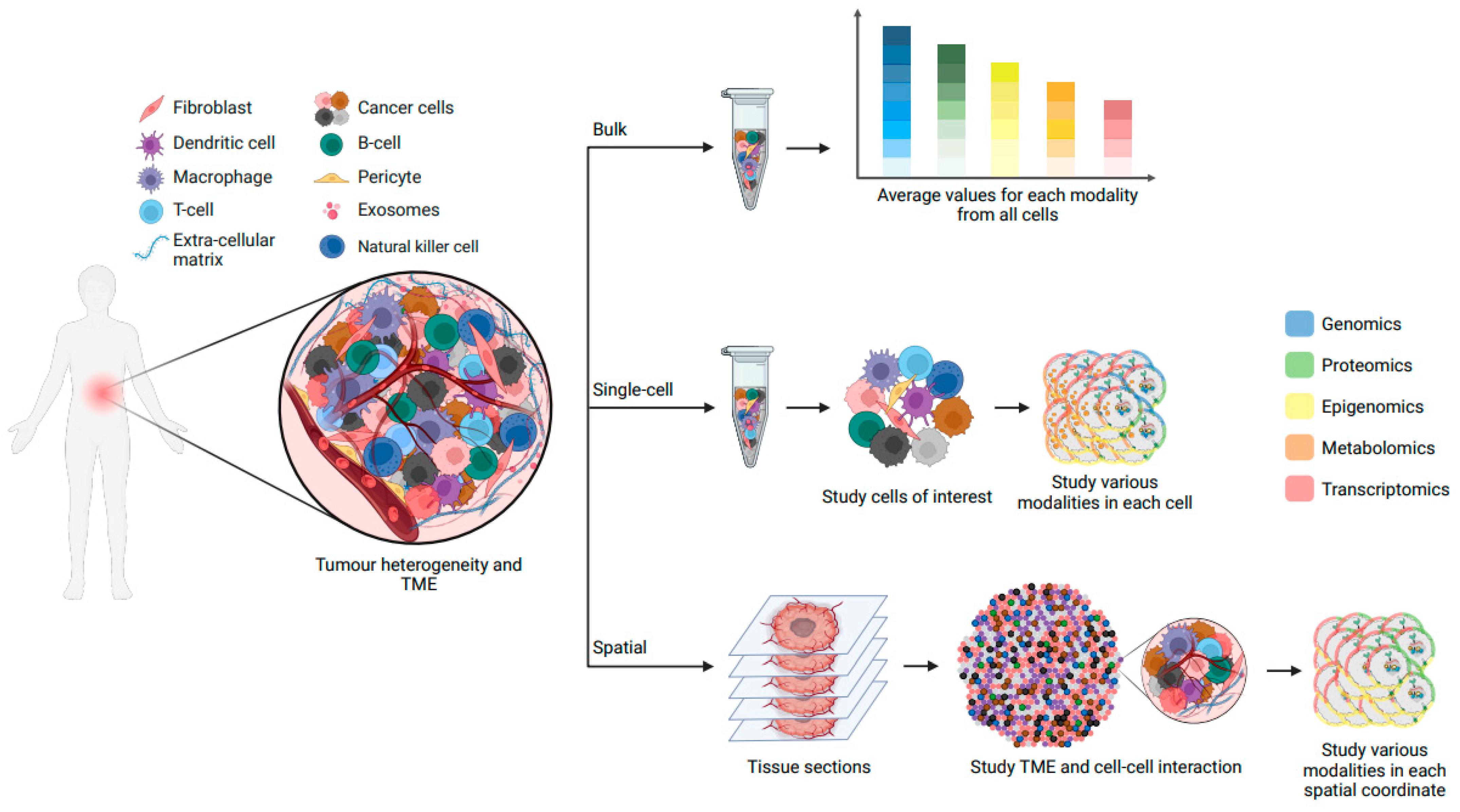
5. Integrating Single-Cell and Spatial Multiomics for Comprehensive Insights into Cancer Evolution
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Yates, L.R.; Campbell, P.J. Evolution of the Cancer Genome. Nat. Rev. Genet. 2012, 13, 795–806. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The Causes and Consequences of Genetic Heterogeneity in Cancer Evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Nam, A.S.; Chaligne, R.; Landau, D.A. Integrating Genetic and Non-Genetic Determinants of Cancer Evolution by Single-Cell Multi-Omics. Nat. Rev. Genet. 2021, 22, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Black, J.R.M.; McGranahan, N. Genetic and Non-Genetic Clonal Diversity in Cancer Evolution. Nat. Rev. Cancer 2021, 21, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Boveri, T. Concerning the Origin of Malignant Tumours by Theodor Boveri. Translated and Annotated by Henry Harris. J. Cell Sci. 2008, 121 (Suppl. S1), 1–84. [Google Scholar] [CrossRef]
- Armitage, P.; Doll, R. The Age Distribution of Cancer and a Multi-Stage Theory of Carcinogenesis. Br. J. Cancer 1954, 8, 1–12. [Google Scholar] [CrossRef]
- Knudson, A.G. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Darwin, C.; Murray, J.; William Clowes and Sons; Evans, B. On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life; John Murray, Albemarle Street: London, UK, 1859; pp. 1–556. [Google Scholar]
- Nowell, P.C. The Clonal Evolution of Tumor Cell Populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Harris, J.F.; Chambers, A.F.; Hill, R.P.; Ling, V. Metastatic Variants Are Generated Spontaneously at a High Rate in Mouse KHT Tumor. Proc. Natl. Acad. Sci. USA 1982, 79, 5547–5551. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.L.; Gatenby, R.A. An Evolutionary Model for Initiation, Promotion, and Progression in Carcinogenesis. Int. J. Oncol. 2008, 32, 729–737. [Google Scholar] [PubMed]
- Vendramin, R.; Litchfield, K.; Swanton, C. Cancer Evolution: Darwin and Beyond. EMBO J. 2021, 40, e108389. [Google Scholar] [CrossRef] [PubMed]
- Michor, F.; Iwasa, Y.; Nowak, M.A. Dynamics of Cancer Progression. Nat. Rev. Cancer 2004, 4, 197–205. [Google Scholar] [CrossRef]
- Gerlinger, M.; Swanton, C. How Darwinian Models Inform Therapeutic Failure Initiated by Clonal Heterogeneity in Cancer Medicine. Br. J. Cancer 2010, 103, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Verduzco, D.; Gatenby, R.A. Evolutionary Dynamics of Carcinogenesis and Why Targeted Therapy Does Not Work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Hadi, K.; Yao, X.; Behr, J.M.; Deshpande, A.; Xanthopoulakis, C.; Tian, H.; Kudman, S.; Rosiene, J.; Darmofal, M.; DeRose, J.; et al. Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs. Cell 2020, 183, 197–210.e32. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang Model of Human Colorectal Tumor Growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.-B.; Brown, A.M.K.; Kim, J.C.; et al. A Renewed Model of Pancreatic Cancer Evolution Based on Genomic Rearrangement Patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal Oncogene Amplification Drives Tumour Evolution and Genetic Heterogeneity. Nature 2017, 543, 122–125. [Google Scholar] [CrossRef] [PubMed]
- deCarvalho, A.C.; Kim, H.; Poisson, L.M.; Winn, M.E.; Mueller, C.; Cherba, D.; Koeman, J.; Seth, S.; Protopopov, A.; Felicella, M.; et al. Discordant Inheritance of Chromosomal and Extrachromosomal DNA Elements Contributes to Dynamic Disease Evolution in Glioblastoma. Nat. Genet. 2018, 50, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Bafna, V.; Mischel, P.S. Extrachromosomal Oncogene Amplification in Tumour Pathogenesis and Evolution. Nat. Rev. Cancer 2019, 19, 283–288. [Google Scholar] [CrossRef]
- Keshavarzian, T.; Lupien, M. ecDNAs Personify Cancer Gangsters. Mol. Cell 2022, 82, 500–502. [Google Scholar] [CrossRef]
- Hung, K.L.; Yost, K.E.; Xie, L.; Shi, Q.; Helmsauer, K.; Luebeck, J.; Schöpflin, R.; Lange, J.T.; Chamorro González, R.; Weiser, N.E.; et al. ecDNA Hubs Drive Cooperative Intermolecular Oncogene Expression. Nature 2021, 600, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Yi, E.; Gujar, A.D.; Guthrie, M.; Kim, H.; Zhao, D.; Johnson, K.C.; Amin, S.B.; Costa, M.L.; Yu, Q.; Das, S.; et al. Live-Cell Imaging Shows Uneven Segregation of Extrachromosomal DNA Elements and Transcriptionally Active Extrachromosomal DNA Hubs in Cancer. Cancer Discov. 2022, 12, 468–483. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef]
- Pérez-González, A.; Bévant, K.; Blanpain, C. Cancer Cell Plasticity during Tumor Progression, Metastasis and Response to Therapy. Nat. Cancer 2023, 4, 1063–1082. [Google Scholar] [CrossRef]
- Fennell, K.A.; Vassiliadis, D.; Lam, E.Y.N.; Martelotto, L.G.; Balic, J.J.; Hollizeck, S.; Weber, T.S.; Semple, T.; Wang, Q.; Miles, D.C.; et al. Non-Genetic Determinants of Malignant Clonal Fitness at Single-Cell Resolution. Nature 2022, 601, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Prieto, T.; Landau, D.A. A Heritable, Non-Genetic Road to Cancer Evolution. Nature 2022, 601, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Chaligne, R.; Gaiti, F.; Silverbush, D.; Schiffman, J.S.; Weisman, H.R.; Kluegel, L.; Gritsch, S.; Deochand, S.D.; Gonzalez Castro, L.N.; Richman, A.R.; et al. Epigenetic Encoding, Heritability and Plasticity of Glioma Transcriptional Cell States. Nat. Genet. 2021, 53, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Joyce, J.A. The Evolving Tumor Microenvironment: From Cancer Initiation to Metastatic Outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Kakiuchi, N.; Yoshida, K.; Sakurai, T.; Kataoka, T.R.; Kondoh, E.; Chigusa, Y.; Kawai, M.; Sawada, M.; Inoue, T.; et al. Evolutionary Histories of Breast Cancer and Related Clones. Nature 2023, 620, 607–614. [Google Scholar] [CrossRef]
- Davis, A.; Gao, R.; Navin, N. Tumor Evolution: Linear, Branching, Neutral or Punctuated? Biochim. Biophys. Acta (BBA)—Rev. Cancer 2017, 1867, 151–161. [Google Scholar] [CrossRef]
- Campbell, L.L.; Polyak, K. Breast Tumor Heterogeneity: Cancer Stem Cells or Clonal Evolution? Cell Cycle 2007, 6, 2332–2338. [Google Scholar] [CrossRef]
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; et al. Mutational Evolution in a Lobular Breast Tumour Profiled at Single Nucleotide Resolution. Nature 2009, 461, 809–813. [Google Scholar] [CrossRef]
- Swanton, C. Intratumor Heterogeneity: Evolution through Space and Time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of Neutral Tumor Evolution across Cancer Types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef]
- Cross, W.C.; Graham, T.A.; Wright, N.A. New Paradigms in Clonal Evolution: Punctuated Equilibrium in Cancer. J. Pathol. 2016, 240, 126–136. [Google Scholar] [CrossRef]
- Korbel, J.O.; Campbell, P.J. Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef]
- Grosberg, R.K.; Strathmann, R.R. The Evolution of Multicellularity: A Minor Major Transition? Annu. Rev. Ecol. Evol. Syst. 2007, 38, 621–654. [Google Scholar] [CrossRef]
- Hammerschmidt, K.; Rose, C.J.; Kerr, B.; Rainey, P.B. Life Cycles, Fitness Decoupling and the Evolution of Multicellularity. Nature 2014, 515, 75–79. [Google Scholar] [CrossRef]
- Du, Q.; Kawabe, Y.; Schilde, C.; Chen, Z.; Schaap, P. The Evolution of Aggregative Multicellularity and Cell–Cell Communication in the Dictyostelia. J. Mol. Biol. 2015, 427, 3722–3733. [Google Scholar] [CrossRef] [PubMed]
- Colizzi, E.S.; Vroomans, R.M.; Merks, R.M. Evolution of Multicellularity by Collective Integration of Spatial Information. eLife 2020, 9, e56349. [Google Scholar] [CrossRef] [PubMed]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. How the Evolution of Multicellularity Set the Stage for Cancer. Br. J. Cancer 2018, 118, 145–152. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, F.; Xing, K.; He, X. The Reverse Evolution from Multicellularity to Unicellularity during Carcinogenesis. Nat. Commun. 2015, 6, 6367. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Lošo, T.; Tautz, D. Phylostratigraphic Tracking of Cancer Genes Suggests a Link to the Emergence of Multicellularity in Metazoa. BMC Biol. 2010, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.C.W.; Lineweaver, C.H. Cancer Tumors as Metazoa 1.0: Tapping Genes of Ancient Ancestors. Phys. Biol. 2011, 8, 015001. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Nam, A.S.; Dusaj, N.; Izzo, F.; Murali, R.; Myers, R.M.; Mouhieddine, T.H.; Sotelo, J.; Benbarche, S.; Waarts, M.; Gaiti, F.; et al. Single-Cell Multi-Omics of Human Clonal Hematopoiesis Reveals That DNMT3A R882 Mutations Perturb Early Progenitor States through Selective Hypomethylation. Nat. Genet. 2022, 54, 1514–1526. [Google Scholar] [CrossRef] [PubMed]
- Cortés-López, M.; Chamely, P.; Hawkins, A.G.; Stanley, R.F.; Swett, A.D.; Ganesan, S.; Mouhieddine, T.H.; Dai, X.; Kluegel, L.; Chen, C.; et al. Single-Cell Multi-Omics Defines the Cell-Type-Specific Impact of Splicing Aberrations in Human Hematopoietic Clonal Outgrowths. Cell Stem Cell 2023, 30, 1262–1281.e8. [Google Scholar] [CrossRef]
- Colom Díaz, P.A.; Mistry, J.J.; Trowbridge, J.J. Hematopoietic Stem Cell Aging and Leukemia Transformation. Blood 2023, 142, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Niroula, A.; Sekar, A.; Murakami, M.A.; Trinder, M.; Agrawal, M.; Wong, W.J.; Bick, A.G.; Uddin, M.M.; Gibson, C.J.; Griffin, G.K.; et al. Distinction of Lymphoid and Myeloid Clonal Hematopoiesis. Nat. Med. 2021, 27, 1921–1927. [Google Scholar] [CrossRef]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited Causes of Clonal Haematopoiesis in 97,691 Whole Genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High Burden and Pervasive Positive Selection of Somatic Mutations in Normal Human Skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Watson, C.J.; Blundell, J.R. Mutation Rates and Fitness Consequences of Mosaic Chromosomal Alterations in Blood. Nat. Genet. 2023, 1–9. [Google Scholar] [CrossRef]
- Brown, D.W.; Cato, L.D.; Zhao, Y.; Nandakumar, S.K.; Bao, E.L.; Gardner, E.J.; Hubbard, A.K.; DePaulis, A.; Rehling, T.; Song, L.; et al. Shared and Distinct Genetic Etiologies for Different Types of Clonal Hematopoiesis. Nat. Commun. 2023, 14, 5536. [Google Scholar] [CrossRef]
- Cagan, A.; Baez-Ortega, A.; Brzozowska, N.; Abascal, F.; Coorens, T.H.H.; Sanders, M.A.; Lawson, A.R.J.; Harvey, L.M.R.; Bhosle, S.; Jones, D.; et al. Somatic Mutation Rates Scale with Lifespan across Mammals. Nature 2022, 604, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Campbell, P.J. Somatic Mutation in Cancer and Normal Cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal Evolution in Breast Cancer Revealed by Single Nucleus Genome Sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.-C.; Dawson, S.-J.; Dawson, M.A. Non-Genetic Mechanisms of Therapeutic Resistance in Cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Li, Z.; Seehawer, M.; Polyak, K. Untangling the Web of Intratumour Heterogeneity. Nat. Cell Biol. 2022, 24, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving Genetic Heterogeneity in Cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- Marjanovic, N.D.; Hofree, M.; Chan, J.E.; Canner, D.; Wu, K.; Trakala, M.; Hartmann, G.G.; Smith, O.C.; Kim, J.Y.; Evans, K.V.; et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 2020, 38, 229–246.e13. [Google Scholar] [CrossRef]
- Tang, F.; Xu, D.; Wang, S.; Wong, C.K.; Martinez-Fundichely, A.; Lee, C.J.; Cohen, S.; Park, J.; Hill, C.E.; Eng, K.; et al. Chromatin Profiles Classify Castration-Resistant Prostate Cancers Suggesting Therapeutic Targets. Science 2022, 376, eabe1505. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare Cell Variability and Drug-Induced Reprogramming as a Mode of Cancer Drug Resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.C.; Krishnamurthy Radhakrishna, V.; et al. Tumor Cells Can Follow Distinct Evolutionary Paths to Become Resistant to Epidermal Growth Factor Receptor Inhibition. Nat. Med. 2016, 22, 262. [Google Scholar] [CrossRef]
- Dhimolea, E.; de Matos Simoes, R.; Kansara, D.; Al’Khafaji, A.; Bouyssou, J.; Weng, X.; Sharma, S.; Raja, J.; Awate, P.; Shirasaki, R.; et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell 2021, 39, 240–256.e11. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Singh Mer, A.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e21. [Google Scholar] [CrossRef]
- Duy, C.; Li, M.; Teater, M.; Meydan, C.; Garrett-Bakelman, F.E.; Lee, T.C.; Chin, C.R.; Durmaz, C.; Kawabata, K.C.; Dhimolea, E.; et al. Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence. Cancer Discov. 2021, 11, 1542–1561. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K. Epigenetics Make Transient States of Cancer Therapy Resistance Permanent. Sci. Transl. Med. 2017, 9, eaan6729. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Emert, B.L.; Reyes Hueros, R.A.; Cote, C.; Harmange, G.; Schaff, D.L.; Sizemore, A.E.; Gupte, R.; Torre, E.; Singh, A.; et al. Memory Sequencing Reveals Heritable Single-Cell Gene Expression Programs Associated with Distinct Cellular Behaviors. Cell 2020, 182, 947–959.e17. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef]
- Davies, A.; Zoubeidi, A.; Beltran, H.; Selth, L.A. The Transcriptional and Epigenetic Landscape of Cancer Cell Lineage Plasticity. Cancer Discov. 2023, 13, 1771–1788. [Google Scholar] [CrossRef] [PubMed]
- Gaiti, F.; Chaligne, R.; Gu, H.; Brand, R.M.; Kothen-Hill, S.; Schulman, R.C.; Grigorev, K.; Risso, D.; Kim, K.-T.; Pastore, A.; et al. Epigenetic Evolution and Lineage Histories of Chronic Lymphocytic Leukaemia. Nature 2019, 569, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Clement, K.; Ziller, M.J.; Boyle, P.; Fan, J.; Gu, H.; Stevenson, K.; Sougnez, C.; Wang, L.; Li, S.; et al. Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia. Cancer Cell 2014, 26, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.; Assenov, Y.; Minner, S.; Bogatyrova, O.; Simon, R.; Koop, C.; Oakes, C.; Zucknick, M.; Lipka, D.B.; Weischenfeldt, J.; et al. Intratumor DNA Methylation Heterogeneity Reflects Clonal Evolution in Aggressive Prostate Cancer. Cell Rep. 2014, 8, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Shipony, Z.; Mukamel, Z.; Cohen, N.M.; Landan, G.; Chomsky, E.; Zeliger, S.R.; Fried, Y.C.; Ainbinder, E.; Friedman, N.; Tanay, A. Dynamic and Static Maintenance of Epigenetic Memory in Pluripotent and Somatic Cells. Nature 2014, 513, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Landan, G.; Cohen, N.M.; Mukamel, Z.; Bar, A.; Molchadsky, A.; Brosh, R.; Horn-Saban, S.; Zalcenstein, D.A.; Goldfinger, N.; Zundelevich, A.; et al. Epigenetic Polymorphism and the Stochastic Formation of Differentially Methylated Regions in Normal and Cancerous Tissues. Nat. Genet. 2012, 44, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased Methylation Variation in Epigenetic Domains across Cancer Types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef]
- Cleal, K.; Norris, K.; Baird, D. Telomere Length Dynamics and the Evolution of Cancer Genome Architecture. Int. J. Mol. Sci. 2018, 19, 482. [Google Scholar] [CrossRef]
- Russo, G.; Tramontano, A.; Iodice, I.; Chiariotti, L.; Pezone, A. Epigenome Chaos: Stochastic and Deterministic DNA Methylation Events Drive Cancer Evolution. Cancers 2021, 13, 1800. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Michalak, E.M.; Burr, M.L.; Bannister, A.J.; Dawson, M.A. The Roles of DNA, RNA and Histone Methylation in Ageing and Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.-T.; Yang, Y.-M.; Sun, M.-M.; He, Y.; Liao, L.; Chen, K.-S.; Li, B. New Insights into the Interplay between Long Non-Coding RNAs and RNA-Binding Proteins in Cancer. Cancer Commun. 2022, 42, 117–140. [Google Scholar] [CrossRef]
- Ashouri, A.; Sayin, V.I.; Van den Eynden, J.; Singh, S.X.; Papagiannakopoulos, T.; Larsson, E. Pan-Cancer Transcriptomic Analysis Associates Long Non-Coding RNAs with Key Mutational Driver Events. Nat. Commun. 2016, 7, 13197. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Sunkel, B.D.; Ray, W.C.; Stanton, B.Z. Chromatin Structure in Cancer. BMC Mol. Cell Biol. 2022, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef]
- Shen, L.; Kondo, Y.; Rosner, G.L.; Xiao, L.; Hernandez, N.S.; Vilaythong, J.; Houlihan, P.S.; Krouse, R.S.; Prasad, A.R.; Einspahr, J.G.; et al. MGMT Promoter Methylation and Field Defect in Sporadic Colorectal Cancer. J. Natl. Cancer Inst. 2005, 97, 1330–1338. [Google Scholar] [CrossRef]
- Al Bakir, M.; Huebner, A.; Martínez-Ruiz, C.; Grigoriadis, K.; Watkins, T.B.K.; Pich, O.; Moore, D.A.; Veeriah, S.; Ward, S.; Laycock, J.; et al. The Evolution of Non-Small Cell Lung Cancer Metastases in TRACERx. Nature 2023, 616, 534–542. [Google Scholar] [CrossRef]
- Frankell, A.M.; Dietzen, M.; Al Bakir, M.; Lim, E.L.; Karasaki, T.; Ward, S.; Veeriah, S.; Colliver, E.; Huebner, A.; Bunkum, A.; et al. The Evolution of Lung Cancer and Impact of Subclonal Selection in TRACERx. Nature 2023, 616, 525–533. [Google Scholar] [CrossRef]
- Coates, J.T.; Sun, S.; Leshchiner, I.; Thimmiah, N.; Martin, E.E.; McLoughlin, D.; Danysh, B.P.; Slowik, K.; Jacobs, R.A.; Rhrissorrakrai, K.; et al. Parallel Genomic Alterations of Antigen and Payload Targets Mediate Polyclonal Acquired Clinical Resistance to Sacituzumab Govitecan in Triple-Negative Breast Cancer. Cancer Discov. 2021, 11, 2436–2445. [Google Scholar] [CrossRef]
- Hua, X.; Zhao, W.; Pesatori, A.C.; Consonni, D.; Caporaso, N.E.; Zhang, T.; Zhu, B.; Wang, M.; Jones, K.; Hicks, B.; et al. Genetic and Epigenetic Intratumor Heterogeneity Impacts Prognosis of Lung Adenocarcinoma. Nat. Commun. 2020, 11, 2459. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Poeta, M.L.; Costantini, M.; Zhang, T.; Shi, J.; Sentinelli, S.; Zhao, W.; Pompeo, V.; Cardelli, M.; Alexandrov, B.S.; et al. The Genomic and Epigenomic Evolutionary History of Papillary Renal Cell Carcinomas. Nat. Commun. 2020, 11, 3096. [Google Scholar] [CrossRef] [PubMed]
- Emert, B.L.; Cote, C.J.; Torre, E.A.; Dardani, I.P.; Jiang, C.L.; Jain, N.; Shaffer, S.M.; Raj, A. Variability within Rare Cell States Enables Multiple Paths toward Drug Resistance. Nat. Biotechnol. 2021, 39, 865–876. [Google Scholar] [CrossRef]
- Goyal, Y.; Busch, G.T.; Pillai, M.; Li, J.; Boe, R.H.; Grody, E.I.; Chelvanambi, M.; Dardani, I.P.; Emert, B.; Bodkin, N.; et al. Diverse Clonal Fates Emerge upon Drug Treatment of Homogeneous Cancer Cells. Nature 2023, 620, 651–659. [Google Scholar] [CrossRef]
- Meir, Z.; Mukamel, Z.; Chomsky, E.; Lifshitz, A.; Tanay, A. Single-Cell Analysis of Clonal Maintenance of Transcriptional and Epigenetic States in Cancer Cells. Nat. Genet. 2020, 52, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Latil, M.; Nassar, D.; Beck, B.; Boumahdi, S.; Wang, L.; Brisebarre, A.; Dubois, C.; Nkusi, E.; Lenglez, S.; Checinska, A.; et al. Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell Stem Cell 2017, 20, 191–204.e5. [Google Scholar] [CrossRef] [PubMed]
- Guilhamon, P.; Chesnelong, C.; Kushida, M.M.; Nikolic, A.; Singhal, D.; MacLeod, G.; Madani Tonekaboni, S.A.; Cavalli, F.M.; Arlidge, C.; Rajakulendran, N.; et al. Single-Cell Chromatin Accessibility Profiling of Glioblastoma Identifies an Invasive Cancer Stem Cell Population Associated with Lower Survival. eLife 2021, 10, e64090. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Mamun, M.A.; Mannoor, K.; Cao, J.; Qadri, F.; Song, X. SOX2 in Cancer Stemness: Tumor Malignancy and Therapeutic Potentials. J. Mol. Cell Biol. 2018, 12, 85–98. [Google Scholar] [CrossRef]
- Mohiuddin, I.S.; Wei, S.-J.; Kang, M.H. Role of OCT4 in Cancer Stem-like Cells and Chemotherapy Resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165432. [Google Scholar] [CrossRef]
- Jeter, C.R.; Yang, T.; Wang, J.; Chao, H.-P.; Tang, D.G. Concise Review: NANOG in Cancer Stem Cells and Tumor Development: An Update and Outstanding Questions. Stem Cells 2015, 33, 2381–2390. [Google Scholar] [CrossRef]
- Chan, J.M.; Zaidi, S.; Love, J.R.; Zhao, J.L.; Setty, M.; Wadosky, K.M.; Gopalan, A.; Choo, Z.-N.; Persad, S.; Choi, J.; et al. Lineage Plasticity in Prostate Cancer Depends on JAK/STAT Inflammatory Signaling. Science 2022, 377, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.E.; Peluffo, G.; Qiu, X.; Temko, D.; Fassl, A.; Li, Z.; Trinh, A.; Seehawer, M.; Jovanović, B.; Alečković, M.; et al. JAK–STAT Signaling in Inflammatory Breast Cancer Enables Chemotherapy-Resistant Cell States. Cancer Res. 2023, 83, 264–284. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Rousseau, B.; Foote, M.B.; Maron, S.B.; Diplas, B.H.; Lu, S.; Argilés, G.; Cercek, A.; Diaz, L.A. The Spectrum of Benefit from Checkpoint Blockade in Hypermutated Tumors. N. Engl. J. Med. 2021, 384, 1168–1170. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; Muyas, F.; Hauck, H.; Smith, O.C.; Sacks, N.J.; Ely, Z.A.; Jaeger, A.M.; Rideout, W.M.; Zhang, D.; Bhutkar, A.; et al. Mismatch Repair Deficiency Is Not Sufficient to Elicit Tumor Immunogenicity. Nat. Genet. 2023, 1–10. [Google Scholar] [CrossRef]
- Ghasemi, S. Cancer’s Epigenetic Drugs: Where Are They in the Cancer Medicines? Pharmacogenom. J. 2020, 20, 367–379. [Google Scholar] [CrossRef]
- Vijayaraghavalu, S.; Dermawan, J.K.; Cheriyath, V.; Labhasetwar, V. Highly Synergistic Effect of Sequential Treatment with Epigenetic and Anticancer Drugs to Overcome Drug Resistance in Breast Cancer Cells Is Mediated via Activation of P21 Gene Expression Leading to G2/M Cycle Arrest. Mol. Pharm. 2013, 10, 337–352. [Google Scholar] [CrossRef]
- Aaltonen, L.A.; Abascal, F.; Abeshouse, A.; Aburatani, H.; Adams, D.J.; Agrawal, N.; Ahn, K.S.; Ahn, S.-M.; Aikata, H.; Akbani, R.; et al. Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A Practical Guide to Single-Cell RNA-Sequencing for Biomedical Research and Clinical Applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef]
- Barkley, D.; Moncada, R.; Pour, M.; Liberman, D.A.; Dryg, I.; Werba, G.; Wang, W.; Baron, M.; Rao, A.; Xia, B.; et al. Cancer Cell States Recur across Tumor Types and Form Specific Interactions with the Tumor Microenvironment. Nat. Genet. 2022, 54, 1192–1201. [Google Scholar] [CrossRef]
- Gavish, A.; Tyler, M.; Greenwald, A.C.; Hoefflin, R.; Simkin, D.; Tschernichovsky, R.; Galili Darnell, N.; Somech, E.; Barbolin, C.; Antman, T.; et al. Hallmarks of Transcriptional Intratumour Heterogeneity across a Thousand Tumours. Nature 2023, 618, 598–606. [Google Scholar] [CrossRef]
- Kinker, G.S.; Greenwald, A.C.; Tal, R.; Orlova, Z.; Cuoco, M.S.; McFarland, J.M.; Warren, A.; Rodman, C.; Roth, J.A.; Bender, S.A.; et al. Pan-Cancer Single-Cell RNA-Seq Identifies Recurring Programs of Cellular Heterogeneity. Nat. Genet. 2020, 52, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Tirosh, I.; Suvà, M.L. Dissecting Human Gliomas by Single-Cell RNA Sequencing. Neuro Oncol. 2018, 20, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-Cell RNA-Seq Supports a Developmental Hierarchy in Human Oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and Oncogenic Programs in H3K27M Gliomas Dissected by Single-Cell RNA-Seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef]
- Richards, L.M.; Whitley, O.K.N.; MacLeod, G.; Cavalli, F.M.G.; Coutinho, F.J.; Jaramillo, J.E.; Svergun, N.; Riverin, M.; Croucher, D.C.; Kushida, M.; et al. Gradient of Developmental and Injury Response Transcriptional States Defines Functional Vulnerabilities Underpinning Glioblastoma Heterogeneity. Nat. Cancer 2021, 2, 157–173. [Google Scholar] [CrossRef]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-Based Classification of Glioblastoma Uncovers a Mitochondrial Subtype with Therapeutic Vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Ogbeide, S.; Giannese, F.; Mincarelli, L.; Macaulay, I.C. Into the Multiverse: Advances in Single-Cell Multiomic Profiling. Trends Genet. 2022, 38, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and Applications for Single-Cell and Spatial Multi-Omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef] [PubMed]
- Baysoy, A.; Bai, Z.; Satija, R.; Fan, R. The Technological Landscape and Applications of Single-Cell Multi-Omics. Nat. Rev. Mol. Cell Biol. 2023, 24, 695–713. [Google Scholar] [CrossRef]
- Johnson, K.C.; Anderson, K.J.; Courtois, E.T.; Gujar, A.D.; Barthel, F.P.; Varn, F.S.; Luo, D.; Seignon, M.; Yi, E.; Kim, H.; et al. Single-Cell Multimodal Glioma Analyses Identify Epigenetic Regulators of Cellular Plasticity and Environmental Stress Response. Nat. Genet. 2021, 53, 1456–1468. [Google Scholar] [CrossRef]
- Gerstberger, S.; Jiang, Q.; Ganesh, K. Metastasis. Cell 2023, 186, 1564–1579. [Google Scholar] [CrossRef]
- Steeg, P.S. Targeting Metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Quinn, J.J.; Jones, M.G.; Okimoto, R.A.; Nanjo, S.; Chan, M.M.; Yosef, N.; Bivona, T.G.; Weissman, J.S. Single-Cell Lineages Reveal the Rates, Routes, and Drivers of Metastasis in Cancer Xenografts. Science 2021, 371, eabc1944. [Google Scholar] [CrossRef]
- Simeonov, K.P.; Byrns, C.N.; Clark, M.L.; Norgard, R.J.; Martin, B.; Stanger, B.Z.; Shendure, J.; McKenna, A.; Lengner, C.J. Single-Cell Lineage Tracing of Metastatic Cancer Reveals Selection of Hybrid EMT States. Cancer Cell 2021, 39, 1150–1162.e9. [Google Scholar] [CrossRef]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic Reprogramming during Pancreatic Cancer Progression Links Anabolic Glucose Metabolism to Distant Metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.-S.; Hwang, C.-I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20. [Google Scholar] [CrossRef] [PubMed]
- LaFave, L.M.; Kartha, V.K.; Ma, S.; Meli, K.; Del Priore, I.; Lareau, C.; Naranjo, S.; Westcott, P.M.K.; Duarte, F.M.; Sankar, V.; et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 2020, 38, 212–228.e13. [Google Scholar] [CrossRef]
- Gutierrez, C.; Al’Khafaji, A.M.; Brenner, E.; Johnson, K.E.; Gohil, S.H.; Lin, Z.; Knisbacher, B.A.; Durrett, R.E.; Li, S.; Parvin, S.; et al. Multifunctional Barcoding with ClonMapper Enables High-Resolution Study of Clonal Dynamics during Tumor Evolution and Treatment. Nat. Cancer 2021, 2, 758–772. [Google Scholar] [CrossRef]
- Umkehrer, C.; Holstein, F.; Formenti, L.; Jude, J.; Froussios, K.; Neumann, T.; Cronin, S.M.; Haas, L.; Lipp, J.J.; Burkard, T.R.; et al. Isolating Live Cell Clones from Barcoded Populations Using CRISPRa-Inducible Reporters. Nat. Biotechnol. 2021, 39, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Seferbekova, Z.; Lomakin, A.; Yates, L.R.; Gerstung, M. Spatial Biology of Cancer Evolution. Nat. Rev. Genet. 2023, 24, 295–313. [Google Scholar] [CrossRef]
- Li, J.; Malouf, C.; Miles, L.A.; Willis, M.B.; Pietras, E.M.; King, K.Y. Chronic Inflammation Can Transform the Fate of Normal and Mutant Hematopoietic Stem Cells. Exp. Hematol. 2023, in press. [CrossRef] [PubMed]
- Rodriguez-Meira, A.; Norfo, R.; Wen, S.; Chédeville, A.L.; Rahman, H.; O’Sullivan, J.; Wang, G.; Louka, E.; Kretzschmar, W.W.; Paterson, A.; et al. Single-Cell Multi-Omics Identifies Chronic Inflammation as a Driver of TP53-Mutant Leukemic Evolution. Nat. Genet. 2023, 55, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, N.A.; Turkalj, S.; Zeng, A.G.X.; Stoilova, B.; Metzner, M.; Nagree, M.S.; Shah, S.; Moore, R.; Usukhbayar, B.; Salazar, M.A.; et al. Selective Advantage of Mutant Stem Cells in Clonal Hematopoiesis Occurs by Attenuating the Deleterious Effects of Inflammation and Aging. bioRxiv 2023. [Google Scholar] [CrossRef]
- Zeng, A.G.X.; Nagree, M.S.; Jakobsen, N.A.; Shah, S.; Murison, A.; Cheong, J.-G.; Lim, I.; Jin, L.; Aguilar-Navarro, A.G.; Araújo, J.; et al. A Hematopoietic Stem Cell Subset That Retains Memory of Prior Inflammatory Stress Accumulates in Aging and Clonal Hematopoiesis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Karimi, E.; Yu, M.W.; Maritan, S.M.; Perus, L.J.M.; Rezanejad, M.; Sorin, M.; Dankner, M.; Fallah, P.; Doré, S.; Zuo, D.; et al. Single-Cell Spatial Immune Landscapes of Primary and Metastatic Brain Tumours. Nature 2023, 614, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cardús, A.; Moran, S.; Musulen, E.; Moutinho, C.; Manzano, J.L.; Martinez-Balibrea, E.; Tierno, M.; Élez, E.; Landolfi, S.; Lorden, P.; et al. Epigenetic Homogeneity within Colorectal Tumors Predicts Shorter Relapse-Free and Overall Survival Times for Patients with Locoregional Cancer. Gastroenterology 2016, 151, 961–972. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial Reconstruction of Single-Cell Gene Expression. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Ravi, V.M.; Will, P.; Kueckelhaus, J.; Sun, N.; Joseph, K.; Salié, H.; Vollmer, L.; Kuliesiute, U.; von Ehr, J.; Benotmane, J.K.; et al. Spatially Resolved Multi-Omics Deciphers Bidirectional Tumor-Host Interdependence in Glioblastoma. Cancer Cell 2022, 40, 639–655.e13. [Google Scholar] [CrossRef]
- Ben-Chetrit, N.; Niu, X.; Swett, A.D.; Sotelo, J.; Jiao, M.S.; Stewart, C.M.; Potenski, C.; Mielinis, P.; Roelli, P.; Stoeckius, M.; et al. Integration of Whole Transcriptome Spatial Profiling with Protein Markers. Nat. Biotechnol. 2023, 41, 788–793. [Google Scholar] [CrossRef]
- Minton, K. Layering Epigenomic and Transcriptomic Space. Nat. Rev. Genet. 2023, 24, 273. [Google Scholar] [CrossRef]
- Zhang, D.; Deng, Y.; Kukanja, P.; Agirre, E.; Bartosovic, M.; Dong, M.; Ma, C.; Ma, S.; Su, G.; Bao, S.; et al. Spatial Epigenome–Transcriptome Co-Profiling of Mammalian Tissues. Nature 2023, 616, 113–122. [Google Scholar] [CrossRef]
- Marsolier, J.; Prompsy, P.; Durand, A.; Lyne, A.-M.; Landragin, C.; Trouchet, A.; Bento, S.T.; Eisele, A.; Foulon, S.; Baudre, L.; et al. H3K27me3 Conditions Chemotolerance in Triple-Negative Breast Cancer. Nat. Genet. 2022, 54, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Cassier, P.A.; Navaridas, R.; Bellina, M.; Rama, N.; Ducarouge, B.; Hernandez-Vargas, H.; Delord, J.-P.; Lengrand, J.; Paradisi, A.; Fattet, L.; et al. Netrin-1 Blockade Inhibits Tumour Growth and EMT Features in Endometrial Cancer. Nature 2023, 620, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Lengrand, J.; Pastushenko, I.; Vanuytven, S.; Song, Y.; Venet, D.; Sarate, R.M.; Bellina, M.; Moers, V.; Boinet, A.; Sifrim, A.; et al. Pharmacological Targeting of Netrin-1 Inhibits EMT in Cancer. Nature 2023, 620, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, L.; Song, Y.; Li, W.; Xu, L. Targeting Macrophages: A Novel Treatment Strategy in Solid Tumors. J. Transl. Med. 2022, 20, 586. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashouri, A.; Zhang, C.; Gaiti, F. Decoding Cancer Evolution: Integrating Genetic and Non-Genetic Insights. Genes 2023, 14, 1856. https://doi.org/10.3390/genes14101856
Ashouri A, Zhang C, Gaiti F. Decoding Cancer Evolution: Integrating Genetic and Non-Genetic Insights. Genes. 2023; 14(10):1856. https://doi.org/10.3390/genes14101856
Chicago/Turabian StyleAshouri, Arghavan, Chufan Zhang, and Federico Gaiti. 2023. "Decoding Cancer Evolution: Integrating Genetic and Non-Genetic Insights" Genes 14, no. 10: 1856. https://doi.org/10.3390/genes14101856
APA StyleAshouri, A., Zhang, C., & Gaiti, F. (2023). Decoding Cancer Evolution: Integrating Genetic and Non-Genetic Insights. Genes, 14(10), 1856. https://doi.org/10.3390/genes14101856