1. Introduction
Microalgae have considerable promise as a production platform for proteins and other high-value molecules. Among eukaryotic microalgae,
Chlamydomonas reinhardtii is the best-studied species and the most amenable to molecular genetic manipulation due to the establishment of efficient transformation methods for all three genomes, expression vectors with inducible and constitutive promoters and multiple selectable marker genes, RNAi, genome editing methods, an indexed insertional mutant library, and other resources [
1,
2,
3]. Already
C. reinhardtii has been engineered to produce many heterologous proteins and small molecules, including industrial enzymes, therapeutic proteins, antibodies, vaccines, nutraceuticals, and small-molecule metabolites [
4,
5,
6,
7,
8].
Expression of heterologous proteins in the
Chlamydomonas chloroplast has a number of advantages compared to expression from the nuclear genome. The chloroplast genome exists in many (~80) copies, transgenes can be targeted to precise locations by homologous recombination, their expression is not silenced by the RNAi machinery as are nuclear transgenes, and fewer proteases reside in the chloroplast than in the cytoplasm [
9,
10]. However, there are some drawbacks to chloroplast expression, including the fact that there are not yet reliable and efficient tools for gene-stacking in this organelle, which is key for expressing multi-protein complexes or entire enzymatic pathways.
Some efforts have been made to fill this gap. One strategy for expressing multiple transgenes in the chloroplast from a single construct is to integrate multiple transgene cassettes in a single transformation event. Noor-Mohammadi et al. did this to achieve expression of three different reporter genes with individual gene expression cassettes in the chloroplast genome of
C. reinhardtii [
11]. Similarly, Larrea-Alvarez et al. integrated a DNA fragment containing three different gene cassettes (and ultimately a fourth in a subsequent transformation) and achieved expression of all transgenic proteins [
12]. This strategy is useful, but the integrated constructs contain three sets of regulatory regions, which increases the size of the construct and also presumably the load on the chloroplast transcription machinery.
An alternative strategy for stacking chloroplast transgenes is polycistronic expression via intercistronic spacer sequences, a method that has been used with success in tobacco and other plants [
13,
14,
15,
16,
17,
18]. To date, there have been two reports relevant to the adaptation of this strategy to
C. reinhardtii. Su et al. integrated the
apcA/apcB operon (encoding the α and β subunits of allophycocyanin) from the cyanobacterium
Spirulina maxima into the
C. reinhardtii chloroplast and were able to detect expression of both proteins by Western blot [
19]. This study served as proof of principle that heterologous proteins could be expressed in
C. reinhardtii using a transgenic, though technically not synthetic, operon. More recently, Macedo-Osorio et al. used a more systematic approach that employed intercistronic expression elements (IEEs) from several endogenous
C. reinhardtii chloroplast operons for which there was published evidence of polycistronic transcription [
20]. These were the spacer sequences that separate the
psbB-psbT,
psbN-psbH,
psaC-petL,
petL-trnN, and
tscA-chIN gene pairs, and they were used in synthetic operons to express
aphA-6 (encodes kanamycin resistance) and
gfp. Transformants with four of their synthetic operons expressed only the first gene in the operon, but lines that harbored the synthetic operons with the
psbN-psbH and
tscA-chlN IEEs produced both aphA-6 protein and GFP. This study represented a breakthrough, as it demonstrated that heterologous proteins could be joined by an IEE/spacer and expressed in the
C. reinhardtii chloroplast from bi-cistronic messages, and it uncovered two functional intercistronic spacer sequences. However, only two heterologous genes were tested, and the successful intercistronic expression elements were relatively large (569 bp and 650 bp).
Here, we set out to extend these synthetic operon efforts by making and testing vectors with much smaller intercistronic spacers and by testing expression of four different genes in either bi- or tri-cistronic operons. We generated constructs containing either the
Synechococcus elongatus apcA/apcB spacer or the
Nicotiana tabacum rps19/rpl22 spacer [
21] or both to connect up to three of the following genes:
C. reinhardtii FBP1 (nuclear gene cDNA that encodes fructose-1, 6-bisphosphatase, or FBPase);
atpB (chloroplast gene, encodes β subunit of
C. reinhardtii chloroplast ATP synthase);
FBA1 (
C. reinhardtii nuclear gene cDNA that encodes fructose-bisphosphate aldolase); and a synthetic gene that encodes a VHH antibody that targets the bacterial food poisoning agent
Campylobacter jejuni. In all, we generated and tested nine constructs, six that contain two-gene operons and three that contain three-gene operons, and we integrated them into the chloroplast genome of strains CC-125 or CC-373. CC-125 is wild type, and CC-373 contains a deletion of the
atpB gene, which is essential for photosynthesis, so the expression of transgenic
atpB could be easily selected. Phenotypic and Western blot analyses indicated that some of the synthetic operons worked to express the intended transgenic proteins, but not all tested genes were expressed.
2. Materials and Methods
2.1. C. reinhardtii Strains, Single-Gene Expression Vectors, and Culture Conditions
C. reinhardtii strains CC-373 and CC-125 were obtained from the
Chlamydomonas Resource Center, University of Minnesota. CC-373, carrying the ac-u-c-2-21 mutation, is a light-sensitive,
atpB deficient strain that has a ~2.5-kb deletion that removes the 3′ part of the
atpB coding region, its 3′ UTR, and part of one of the chloroplast genome inverted repeats [
22]. CC-373 was grown in Tris Acetate Phosphate (TAP) medium (Gorman and Levine, 1965), pH 7.3, in a Percival growth chamber (model#: AR-60L, Percival Scientific, Inc., Perry, IA, USA) at 24 °C in the dark. CC-125 was grown in TAP medium, pH 7.3, in a Percival growth chamber at 24 °C and continuous light (~126 microeinstein per second per square meter, μE m
−2 s
−1).
2.2. Generation of Plasmid Vectors
Plasmid pFBP1 (previously named pWD-CpFBP1) was described in [
23]. Plasmid pFBA1 contains, in place of the
FBP1 sequence in pFBP1, the
C. reinhardtii FBA1 coding sequence (nuclear), gene synthesized (Genscript Biotech Corporation, Piscataway, NJ, USA) to be codon optimized for the
C. reinhardtii chloroplast and to contain a C-terminal myc tag, flanked by SfoI and MscI restriction sites. Plasmid pVHH contains a similarly codon-optimized fragment encoding a VHH antibody targeting the flagellum of the food poisoning agent
Campylobacter jejuni [
24], but also a 38 amino acid N-terminal Streptavidin-Binding Peptide (SBP) and C-terminal myc tag. Plasmid p-423 (contains the
aadA gene conferring spectinomycin resistance [
25]) was obtained from the Chlamydomonas Resource Center (St. Paul, MN, USA).
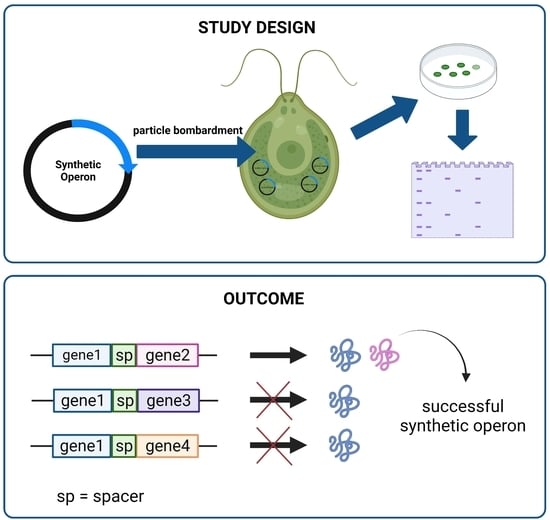
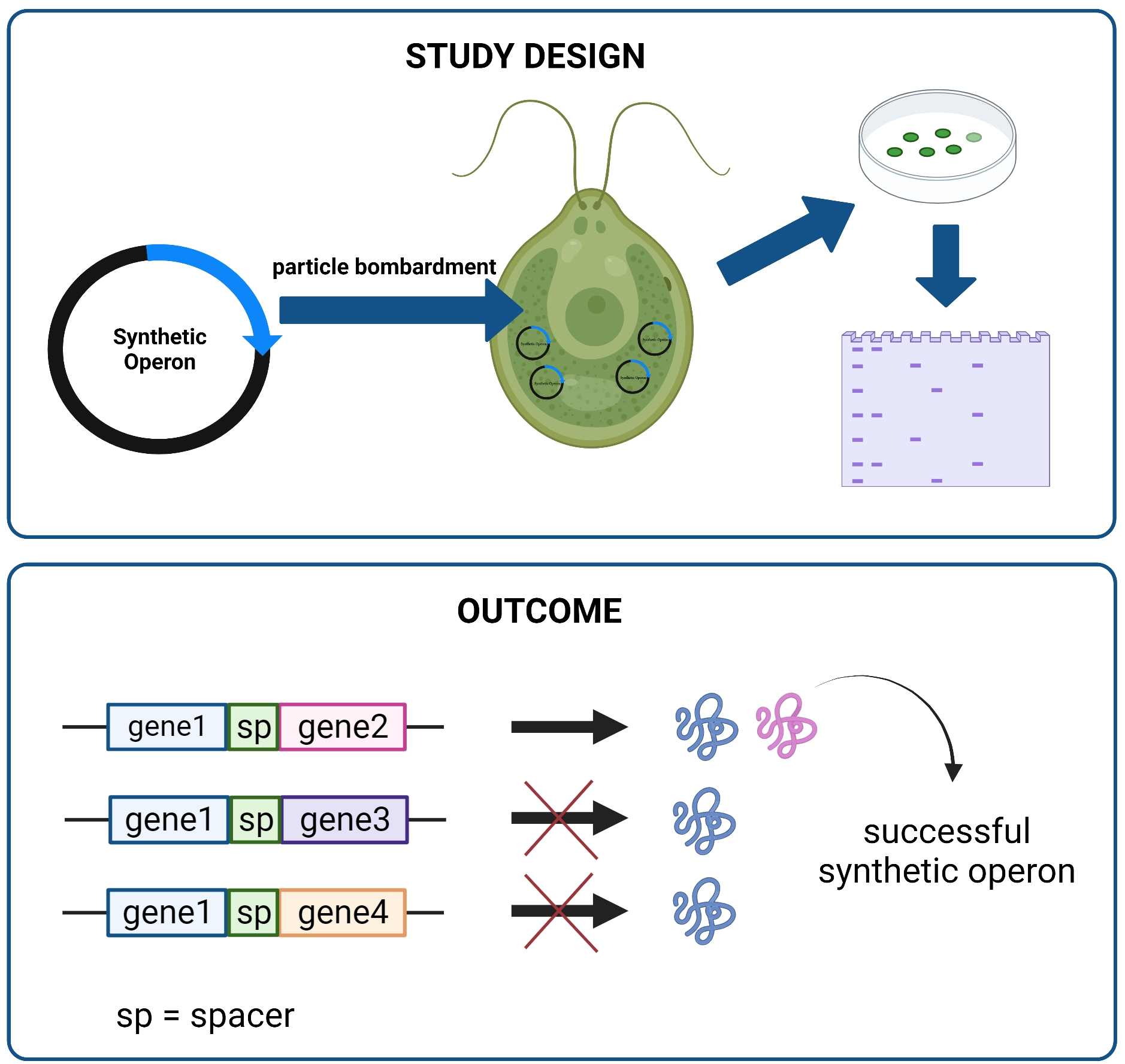
The pFBP1-cs-ts-atpB vector was generated by combining gene-synthesized and existing gene fragments, as described below, and schematic depiction of the strategy for generating this and other synthetic operons is provided in
Figure 1A. A 1614-bp DNA fragment was gene synthesized (Genscript) to contain the 55-bp spacer sequence of the
Synechococcus elongatus apc operon that separates the stop codon of the
apcA gene and the start codon of the
apcB gene and the 47-bp
Nicotiana tabacum rps/
rpl operon spacer that separates the
rps19 stop codon from the
rpl22 start codon, and the 1470-bp
C. reinhardtii atpB coding sequence. The cyanobacterial and tobacco spacer sequences were separated by a 9-bp sequence containing a start codon and SfoI restriction site, and that operon cassette was preceded by SnaBI and PmeI restriction sites (surrounding a stop codon) at the 5′ end to permit joining of either operon spacer with downstream
atpB gene to an upstream gene through blunt-end cloning. The
atpB coding region was synthesized to contain a StuI restriction site immediately following its start codon and an MscI restriction site immediately following its final codon. The gene-synthesis fragment was digested with SnaBI and MscI to liberate a 1608-bp blunt-ended fragment that was ligated into MscI-digested pFBP1 to generate p-FBP1-cs-ts-atpB. p-FBP1-cs-ts-atpB was used to generate the nine chloroplast expression vectors tested in this study, which contain different combinations of the
FBP1,
atpB,
FBA1, and
VHH genes connected by cyanobacterial and/or tobacco chloroplast operon spacers as described below.
pFBP1-cs-atpB and pFBP1-ts-atpB, which contain the FBP1 and atpB genes connected by the cyanobacterial spacer or the tobacco spacer, respectively, were generated as follows. pFBP1-cs-ts-atpB was digested with restriction enzymes SfoI and StuI (Thermo Fisher Scientific, Waltham, MA, USA) and then religated to produce pFBP1-cs-atpB, and it was digested with PmeI and SfoI and then religated to produce pFBP1-ts-atpB.
pFBP1-cs-VHH and pFBP1-ts-VHH, which contain the FBP1 and VHH genes connected by the cyanobacterial spacer or the tobacco spacer, respectively, were generated as follows. First, pFBP1-cs-atpB was digested with SfoI and MscI to remove the fragment containing the atpB gene and cyanobacterial spacer, and the remaining 9.355-kb vector fragment from this digest was ligated with the 1005-bp coding sequence fragment of the VHH gene derived from a Eco47III and MscI digest of vector pVHH; the completed expression vector was named pFBP1-cs-VHH. pFBP1-ts-VHH was generated by ligating the Eco47III-MscI VHH fragment into the vector fragment produced by StuI and MscI digestion of pFBP1-ts-atpB.
pFBP1-cs-FBA and pFBP1-ts-FBA, which contain FBP1 and FBA1 genes connected by the cyanobacterial spacer or the tobacco spacer, respectively, were generated as follows. pFBP1-cs-FBA was produced by ligating the 1170-bp coding sequence fragment of FBA1 that was liberated by digesting pFBA1 with SfoI and MscI into the vector fragment derived from a digest of pFBP1-cs-atpB with StuI and MscI. Similarly, pFBP1-ts-FBA was generated by ligating the 1170-bp SfoI-MscI FBA1 fragment into the vector fragment derived from a StuI + MscI digest of pFBP1-ts-atpB.
Expression vectors pFBP1-cs-FBA-cs-atpB, pFBP1-cs-FBA-ts-atpB, and pFBP1-ts-FBA-ts-atpB were generated as follows. The 1542-bp fragment containing the cyanobacterial spacer and
atpB gene coding sequence was liberated by digesting pFBP1-cs-atpB with PmeI and MscI, then was ligated into the vector fragment derived from an MscI digest of pFBP1-cs-FBA to generate pFBP1-cs-FBA-cs-atpB. Similarly, the 1532-bp SfoI-MscI fragment from pFBP1-ts-atpB was ligated into MscI-digested pFBP1-cs-FBA to produce pFBP1-cs-FBA-ts-atpB. Finally, the 1532-bp SfoI-MscI fragment from p-FBP1-cs-ts-atpB containing the tobacco operon spacer and
atpB coding sequence was ligated into MscI-digested pFBP1-ts-FBA, to generate pFBP1-ts-FBA-ts-atpB. All final constructs were verified by diagnostic restriction digestions and sequencing (primers displayed in
Table 1) to verify expected fragment sizes, to determine that no unintended sequence changes had occurred during cloning steps, and to verify that insert fragments were all in correct orientation.
2.3. C. reinhardtii Chloroplast Transformation
Chloroplast transformation was performed using the biolistic particle bombardment method as described previously [
23]. Strain CC-373 cells were cultured in 25 mL TAP medium with dim light for seven days to a density of 3 × 10
6 cells/mL. The culture was then centrifuged at 3000×
g for 5 min, and the pellet was resuspended in TAP medium to a concentration of 30 × 10
6 cells/mL. In total, 330 µL of the resuspended cells was spread onto a TAP-agar plate supplemented with 100 µg/mL spectinomycin and allowed to dry, uncovered, in a dark sterile hood for 10 min.
Five macrocarriers (Bio-Rad Laboratories, Inc., Hercules, CA, USA) were washed in absolute ethanol and assembled into macrocarrier holders, then placed in a desiccator for 2–3 h to dry. DNA coating of gold particle microcarriers (550 µM diameter; SeaShell Technology, LLC, La Jolla, CA, USA) was as follows: 50 µL of binding buffer was mixed with 10 µL of plasmid, 1 µg/µL, in a sterile 1.5 mL microcentrifuge tube; 60 µL of gold carriers (50 mg/mL) and 100 µL of precipitation buffer were added to the mixture. The mixture was briefly centrifuged for 10 s at 12,000× g, and the supernatant was removed. The pellet was washed with 500 µL cold absolute ethanol, and the supernatant was removed. The pellet was resuspended in 50 µL of absolute ethanol, and 10 µL of DNA-coated microcarriers were loaded onto the inner area of a macrocarrier. The macrocarrier was dried for 10 min in a desiccator.
The plasmid DNA coated with gold particles was bombarded into C. reinhardtii cells via PDS-100/He gene gun (Bio-Rad Laboratories, Inc., USA) using helium gas and a 1350-psi rupture disk. After bombardment, the plates were incubated in a Percival growth chamber with dim light for approximately 3 weeks. Transformant colonies appeared after ~16 days.
For wild-type strain CC-125, the preparation of cells and plasmid DNA coating of microcarriers were carried out as described above. However, CC-125 cells were cultured under continuous light for four days prior to bombardment, after which the plates were incubated in dim light overnight, then placed under continuous light for two weeks.
2.4. Genomic DNA Extraction and Colony PCR
Genomic DNA was extracted using the Chelex method, as described below; 1–5 × 10
6 C. reinhardtii cells from transformant and recipient strains were added to 60 µL of Chelex 100 solution buffer (0.1 M NaOH + 5% Chelex-100 (Bio-Rad) and the mixture vortexed to resuspend the resin, in a microcentrifuge tube. The samples were boiled at 65 °C for 15 min and centrifuged at 11,000×
g for 1 min. In total, 20 µL of the supernatant was transferred into 30 µL of 50 mM TrisCl, pH 8, and stored at −20 °C. The cell lysate was used as a template DNA for PCR. For some samples, the colony PCR method was used, described in [
26]. Briefly, 1 µL of genomic DNA extracts from transformants, and recipient strains (prepared as described above) were inoculated into 19 µL of DreamTaq master mix (Thermo Fisher Scientific, Inc.), primer sets, and dH
2O. Gene-specific forward and reverse primers (Invitrogen) are listed in
Table 1. All PCR were performed using a T100
TM Thermal Cycler (Bio-Rad) for 29 cycles under the following thermal cycle conditions: 98 °C for 30 s, 98 °C for 30 s, 54 °C for 30 s, and 72 °C for 2 min, with a final extension at 72 °C for 10 min. DNAs from transformant cells were amplified with primer sets P1-P5 to screen for the presence of the genes
FBP1,
atpB,
VHH, and
FBA1 in integrated transgene cassettes, respectively.
Table 1.
List of oligonucleotide primers for sequencing and colony PCR.
Table 1.
List of oligonucleotide primers for sequencing and colony PCR.
| Name | Sequence (5′-3′) | Purpose |
|---|
| Syn-op1 | CCAGAAAAAGTTCATCAACGTGTACCTTTATT | Sequencing |
| Syn-op2 | TACACGTTGATGAACTTTTTCTGG | Sequencing |
| Syn-op3 | AGCTTCAACAGGTTCATTTAAAGG | Sequencing |
| P1 (FBP1) | F: GCACAAAGCAGTTCTAGTCC
R: ACCATCAATTGTACCTTTCATCTC | Genomic PCR |
| P2 (atpB) | F: AACATTTTCCGTTTCGTACAAGCT
R: GTCCTGCCAACTGCCTATG | Genomic PCR |
| P3 (VHH) | F: AGGTAGTTATCAATATTGGGGTCA
R: GTCCTGCCAACTGCCTATG | Genomic PCR |
| P4 (FBA1) | F: TACATTAGGTCCAGGTGATTATTC
R: AGGAGCTGTACGGTGAATTGGTAA | Genomic PCR |
| P5 (FBA1/atpB) | F: TACATTAGGTCCAGGTGATTATTC
R: AGGAGCTGTACGGTGAATTGGTAA | Genomic PCR |
2.5. Phenotypic Screening for atpB Transformants
A total of 16 days after bombardment, cells from surviving spectinomycin-resistant colonies were streaked onto acetate-free Tris Phosphate (TP) medium plates (medium titrated with HCl to pH 7.0) supplemented with 100 µg/mL spectinomycin and grown in a Percival growth chamber under continuous light and at 24 °C for 5–7 days to assay for photoautotrophic growth.
2.6. Western Blot Analysis
Western blots to assay accumulation of transgenic protein were performed as described previously [
23] with minor modifications. Cells of transformants were cultured in 25 mL TAP medium (supplemented with 100 µg/mL spectinomycin) to mid-log phase, pelleted by centrifugation at 3000×
g for 5 min, mixed with 200 µL lysis buffer (4% SDS, 100 mM Tris pH 6.8, 10% glycerol, and 100 mM DTT) and boiled for 5 min. Protein extracts from the cells were loaded onto 12% SDS-PAGE gels and electrophoresed at 100 V for 85–100 min. The proteins were transferred to a nitrocellulose membrane (Amersham-Protran; GE Healthcare, Silver Spring, MD, USA) in a Mini Protean Tetra Cell (Bio-Rad) set to 21 V and 50 milliamps. The following day, the membrane was blocked with 5% milk (10% Carnation non-fat) for one hour. To detect FBP1 and VHH proteins, the membrane was probed with a primary anti-FLAG mouse monoclonal antibody (Sigma-Aldrich, Inc., St. Louis, MO, USA) diluted 1:2000 in 2 mL of 1X PBST (0.01% Tween in phosphate-buffered saline) with 0.06 g Bovine Serum Albumin (BSA) (Sigma-Aldrich) for 1 h. The membrane was then probed with a secondary goat anti-mouse IgG antibody-HRP conjugate (diluted 1:40,000 in 40 mL of 5% milk solution) for 30 min. To detect atpB protein, the membrane was reacted with a primary rabbit polyclonal anti-atpB-antibody (Agrisera, Vännäs, Sweden) diluted 1:2000 in 2 mL of 1X PBST with 0.06 g BSA for one hour, then probed with a secondary goat anti-rabbit IgG antibody diluted 1:40,000 in 40 mL 5% milk solution for 30 min. To detect FBA1 protein, membranes were probed with a primary anti-myc mouse monoclonal antibody (Active Motif, Carlsbad, CA, USA) diluted 1:2000 in 2 mL of 1X PBST with 0.06 g BSA for one hour. After that, the membrane was probed with a secondary goat anti-mouse IgG-HRP conjugate antibody diluted at 1:40,000 in 40 mL of 5% milk solution for 30 min. The membrane was washed 5 times with 1X PBST, developed with Super Signal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) for 5 min, and exposed to X-ray film.
4. Discussion
As part of a prior investigation, we reported overexpression of
C. reinhardtii FBPase in the chloroplast using a newly generated expression vector [
23]. Here, with the goal of improving algal chloroplast protein expression technology, we used that vector as a starting point to generate synthetic operons that contain previously untested intercistronic spacer sequences (spacers), one derived from the cyanobacterial species
S. elongatus and the other from the tobacco chloroplast genome. These spacers (
apcA/apcB and
rps19/rpl22), at 56 and 47 bp, respectively, are >10 times smaller than the
C. reinhardtii intergenic expression elements shown recently to work in the first (and thus far only other) synthetic operons reported to function in
C. reinhardtii [
20]. Synthetic operons in which either the cyanobacterial or the tobacco spacer separated the
FBP1 and
atpB genes worked to express both encoded proteins, showing that these short spacers from disparate, heterologous species can function well in
C. reinhardtii chloroplast operons. However, all operons containing
FBP1 and either of two other genes,
C. reinhardtii FBA1 or a gene that encodes a synthetic camelid heavy-chain variable domain (VHH) antibody, expressed only the
FBP1 gene product and not the
FBA1 or
VHH products. This result was surprising given that both FBA1 and VHH protein products were produced at easily detectable levels when either was expressed in a single-gene construct, and it raises the possibility that sequence-specific elements within coding regions of genes, or more generally, the overall nature of that coding sequence, might have strong effects on protein expression from synthetic operons in
C. reinhardtii. It also raises the question of how commonplace it will be for genes in the second position to express well with the
apcA/apcB and
rps19/rpl22 spacers. Future efforts should include testing additional genes in that position, including
FBP1.
We chose to use the
apcA/apcB and
rps19/rpl22 spacers in our synthetic operons because their small size (they are among the shortest intercistronic spacers we could find among published prokaryotic or chloroplast genomes) helped to make the synthetic operons compact and easy to manipulate. Indeed, these spacers are at least 500 bp smaller than the spacers/IEEs used in the previously reported
C. reinhardtii synthetic operons [
20]. Small spacer size should be especially advantageous for efforts to stack three or more cistrons into the same operon. For instance, it might be difficult or impossible to integrate inserts above a certain size into the chloroplast genome. Interestingly these two spacers are not similar at all to each other at the primary sequence level, except that both are fairly A/T rich (64.3–72%), suggesting that the requirements for spacer function in a synthetic operon might be relatively flexible. However, there must be some requirements, at least for some synthetic operons. Macedo-Osorio et al. found that only two of the six endogenous IEEs/intercistronic spacers they tested functioned in their
C. reinhardtii synthetic operon constructs [
20], and Lu et al. found that processing of a synthetic operon primary transcript in tobacco was defective when they used a spacer derived from an intergenic region of the chloroplast instead of an intercistronic spacer from the
psbB operon [
17].
A possible disadvantage of small spacers is that they might not contain regulatory sequences required for the stability of processed versions of some synthetic operon transcripts. At least some polycistronic chloroplast mRNAs in higher plants are initially processed, presumably by endonucleases that cleave A/U-rich regions within the intercistronic spacer, after which those post-cleavage transcripts are believed to be stabilized by pentatricopeptide repeat (PPR) or tetratricopeptide repeat (TPR) proteins that bind within an intercistronic RNA sequence, blocking exonuclease-mediated degradation [
27,
28,
29]. Many
C. reinhardtii chloroplast mRNAs are polycistronic, and some of these are also processed into monocistronic units [
20,
29,
30,
31,
32], though little is known about how these specific processing events are regulated in this species.
Another possible disadvantage of small spacers is that they might not permit the required secondary structure for the unmasking of the ribosome binding site (RBS) in the second cistron of polycistronic mRNAs. In our experiments, the small spacers did work for
atpB, possibly because the sequence of this gene near the RBS is such that the RBS is not sequestered within a double-stranded RNA region, but the secondary structure situation might be very different for the
VHH and
FBA genes. Conceivably even larger spacers, such as the ones used in [
20], might contain sequences that lead to masking of the RBS for some downstream genes, explaining why some of the synthetic operons tested in that study did not work. A follow-up analysis of accumulated transcripts in transformants that did not express the second gene in the synthetic operon could shed some light on this question.
In general, the coding sequence of the downstream gene in a synthetic operon might greatly influence the stability of the processed transcripts. It seems possible that the
FBA1 and
VHH coding sequences lack features required for post-processing stability, which could explain why those genes could be expressed from single-gene vectors but did not work in our synthetic operons, even when they were sandwiched between two cistrons that were translated (
Figure 4). Consistent with this idea, it has been shown that sequences within the coding region of a
C. reinhardtii chloroplast gene can have dramatic effects on transcript accumulation [
33].
Given these considerations, in the future, it might be helpful to use computational methods to predict (and then subsequently alter) transcript structural features when designing synthetic operons. Secondary structure predictions were previously used to guide the modification of a single-gene
C. reinhardtii chloroplast expression vector. In this case, mutating the 5′ UTR to alter predicted stem structures had profound effects on translation [
34]. Another strategy that has promise is to engineer into the spacer sequence the binding site for a known PPR or TPR protein to stabilize the processed transcripts that might arise from the synthetic operon. As a variation of that strategy, PPR or TPR proteins could be engineered to bind and stabilize existing spacer sequences. To that end, a combinatorial amino acid code has been worked out for the helical repeat motifs of some plant PRR proteins, making it possible to design these proteins to have specificity for a wide variety of RNA sequences [
35,
36]. In conclusion, our findings represent an advance in
C. reinhardtii synthetic operon technology, but they also serve as a reminder that gaps in our understanding of chloroplast RNA processing and translation in this species might need to be filled to realize the potential of this method. Further investigation of how intercistronic spacer sequences function, both those native to
C. reinhardtii and those from other species, will likely be necessary to fully harness this approach.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



