
Characterization of the Complete Mitochondrial Genome of a Flea Beetle Luperomorpha xanthodera (Coleoptera: Chrysomelidae: Galerucinae) and Phylogenetic Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Genome Sequencing, Assembly and Annotation
2.3. Mitogenome Sequence Analyses
2.4. Phylogenetic Analysis
3. Results and Discussion
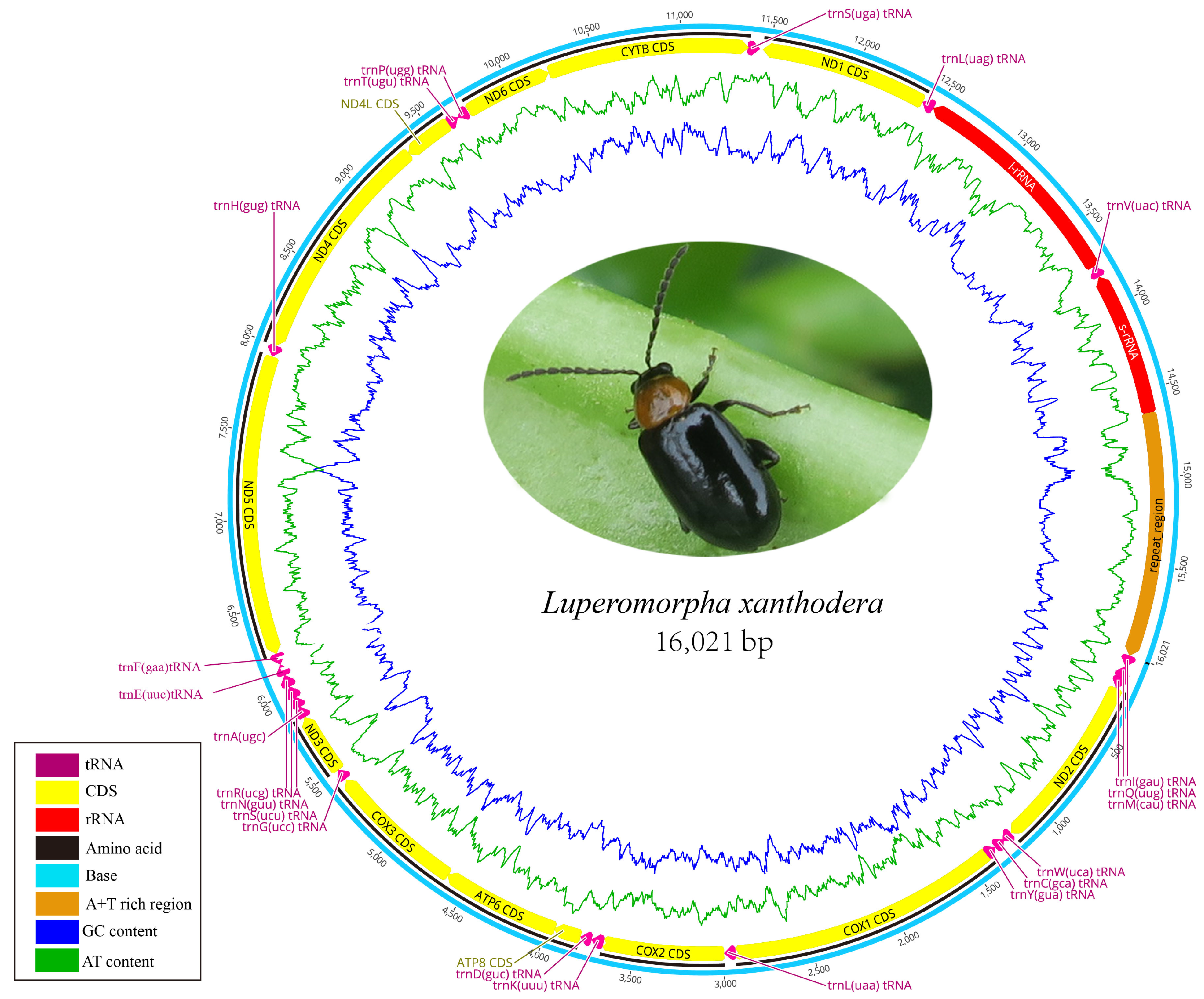
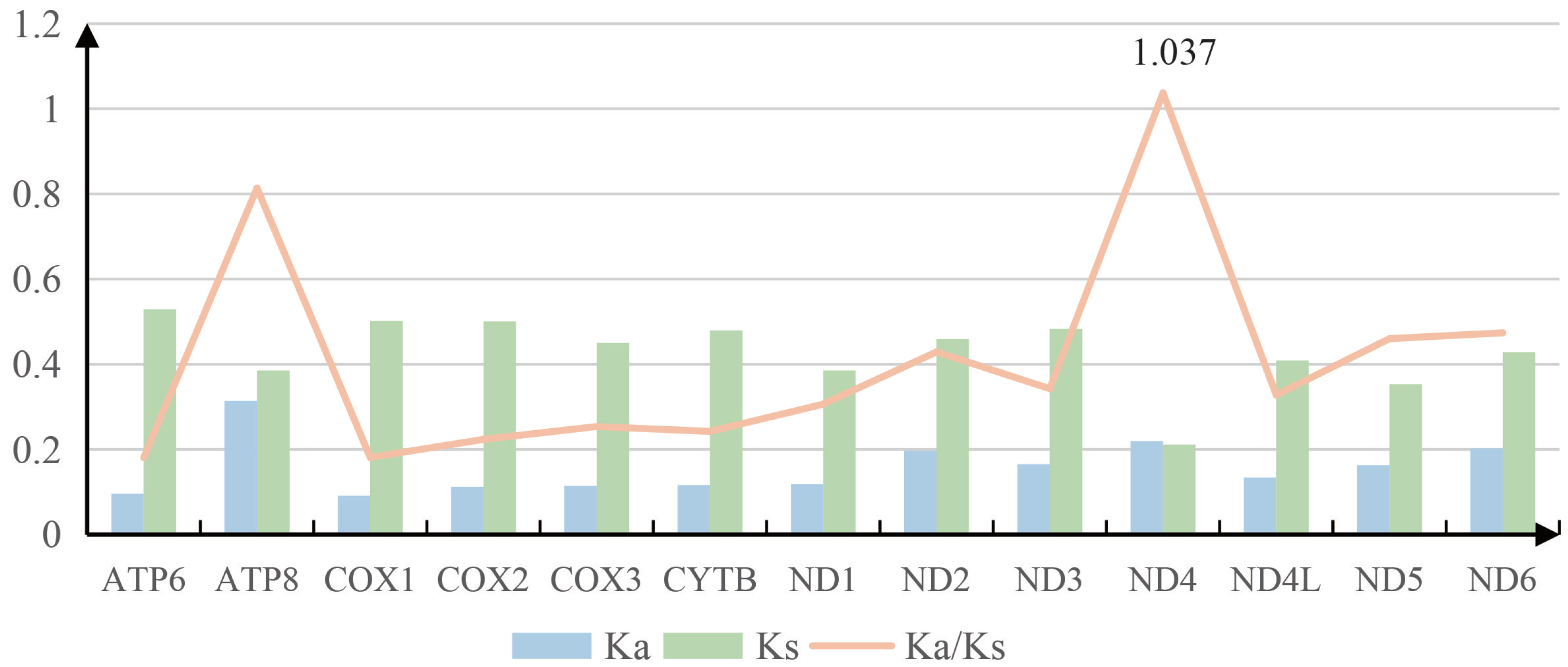
3.1. Mitogenome Organization and Nucleotide Composition
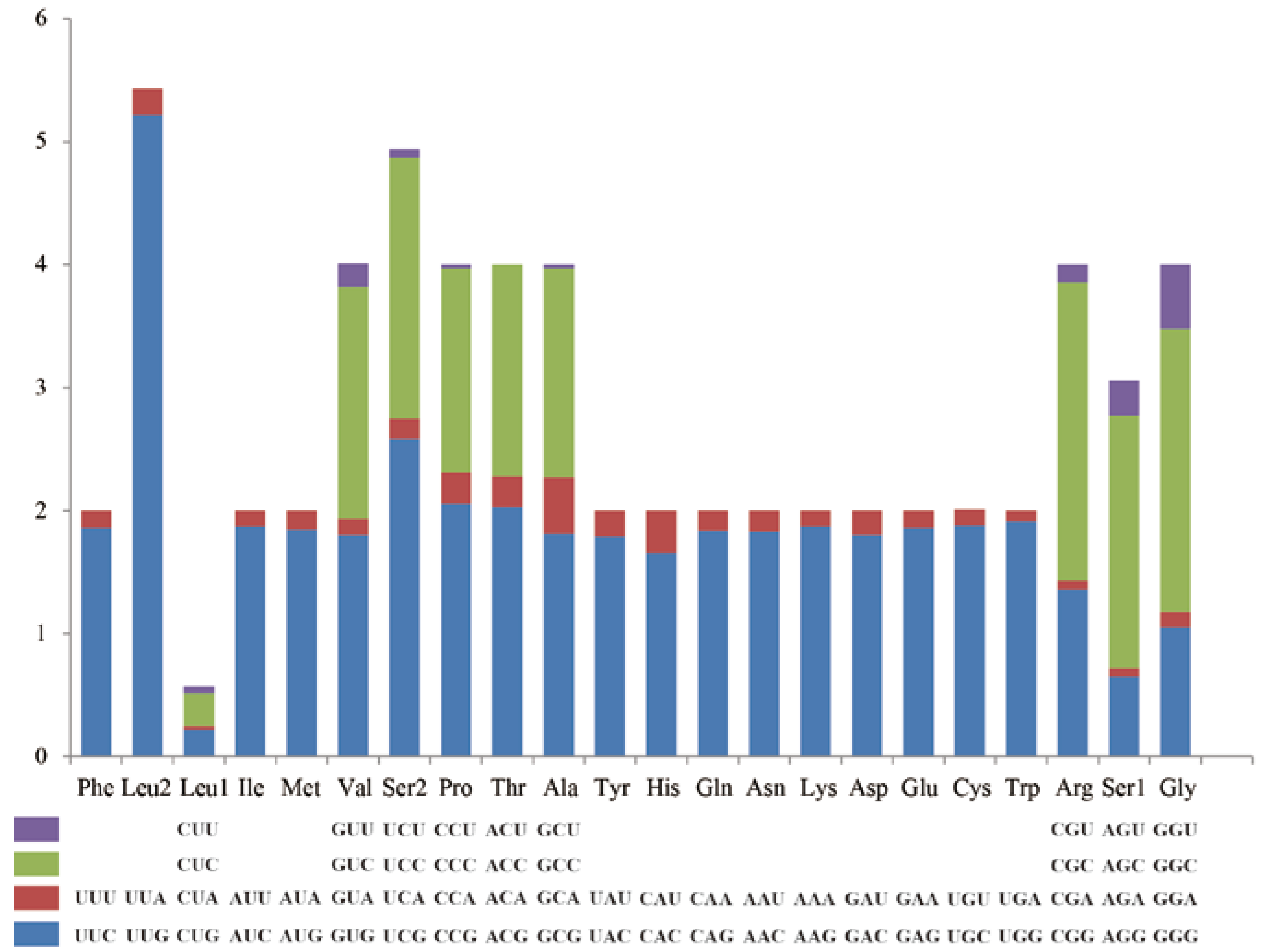
3.2. Protein-Coding Genes and Codon Usage
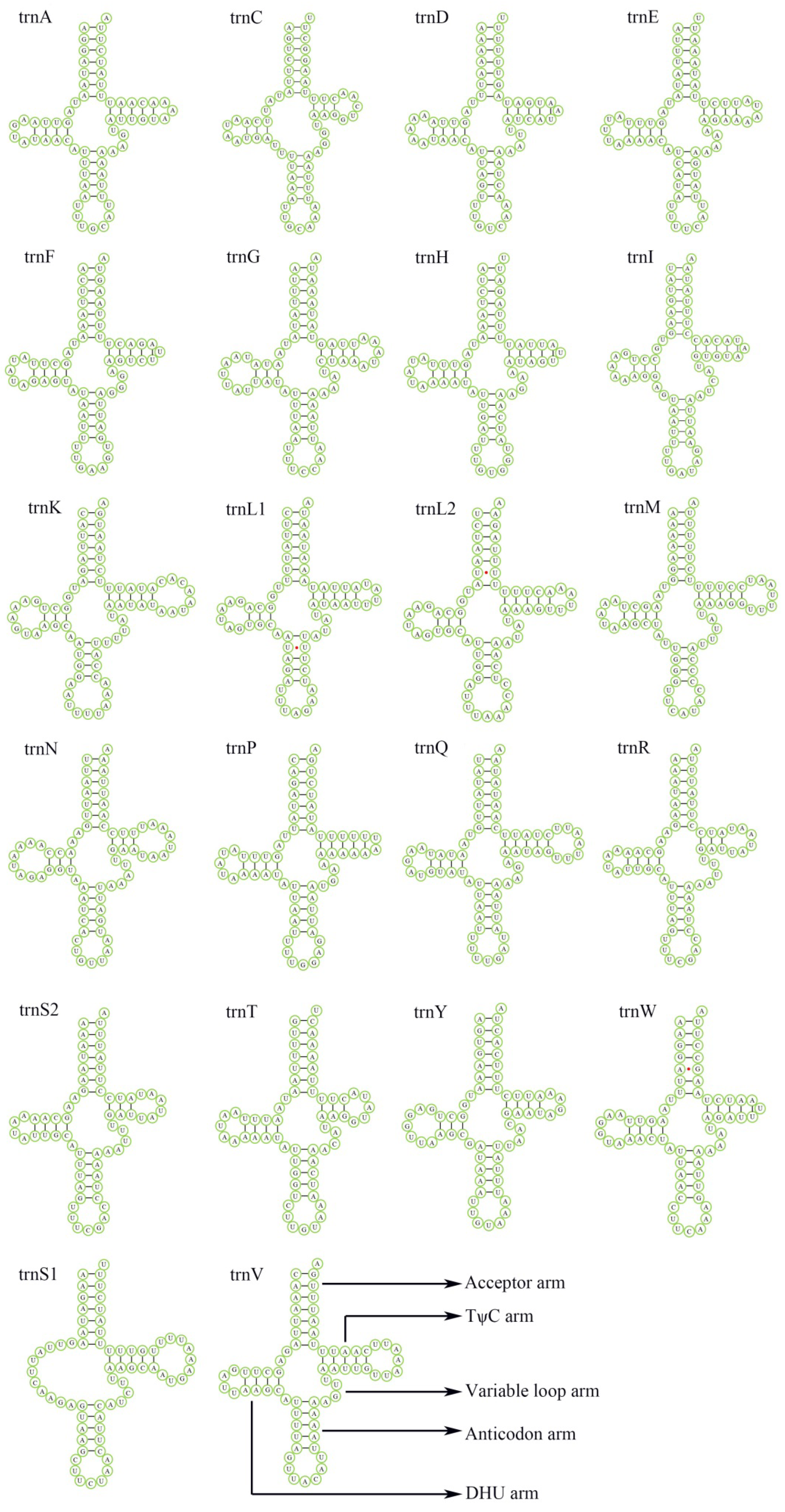
3.3. Transfer and Ribosomal RNA Genes
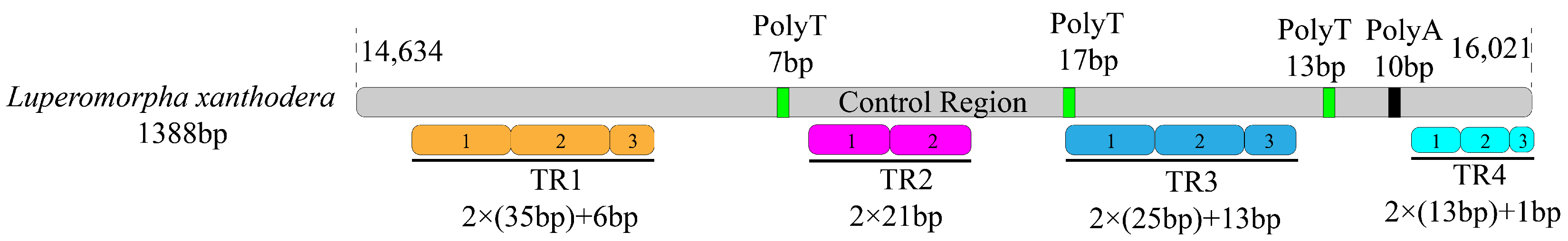
3.4. A + T Rich Region
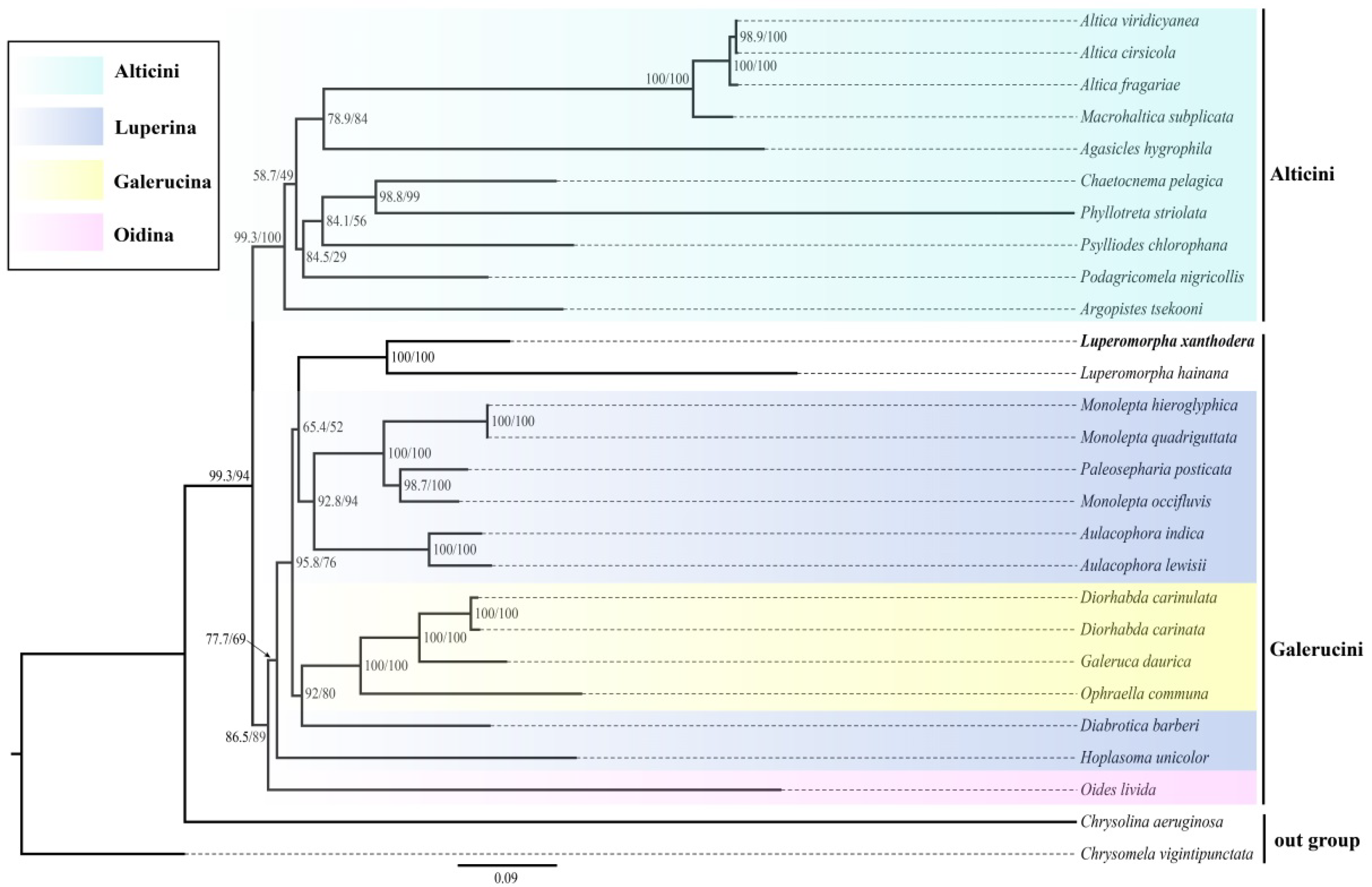
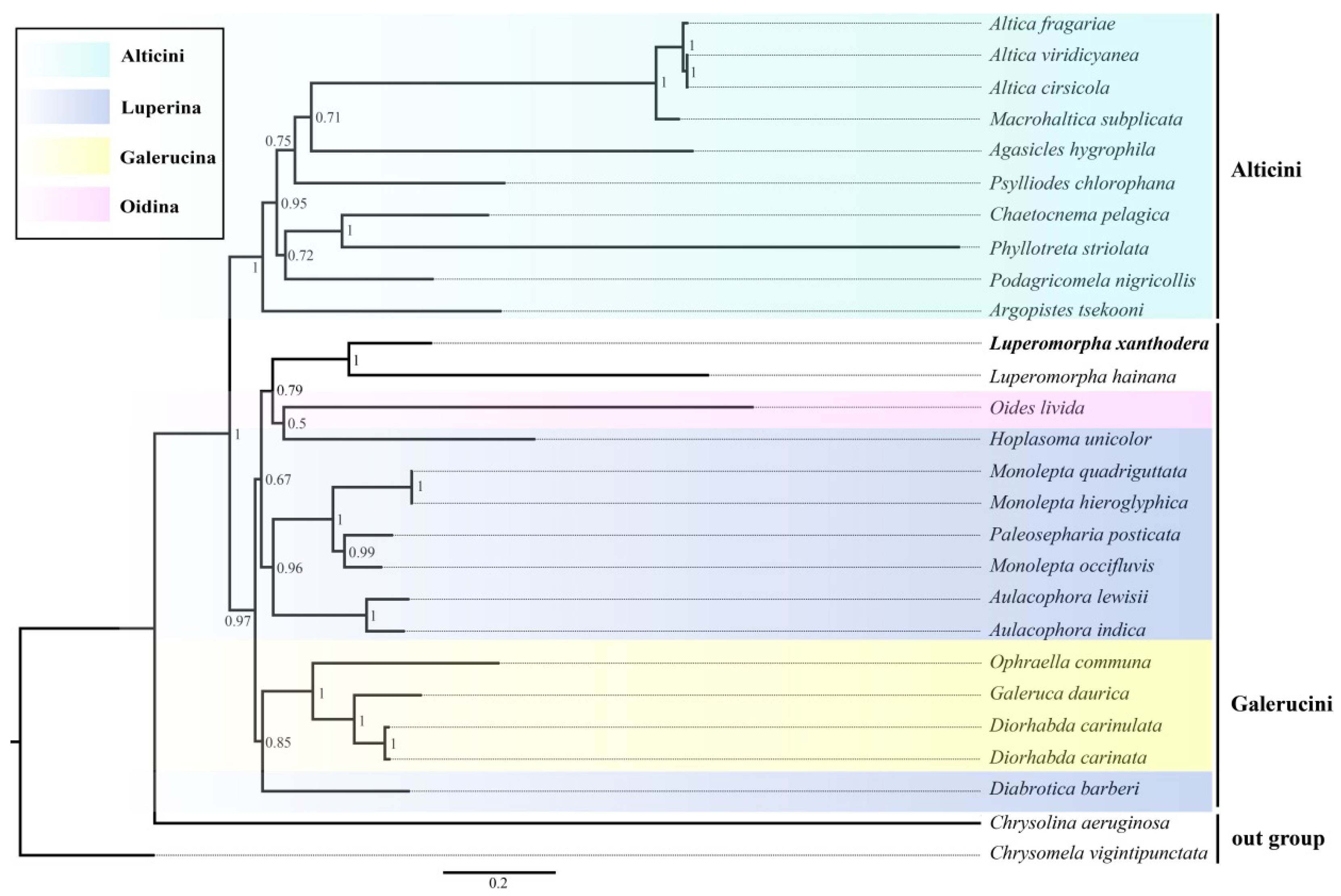
3.5. Phylogenetic Relationships
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhongying, Q.; Huihui, C.; Hao, Y.; Yuan, H.; Huimeng, L.; Xia, L.; Xingchun, G. Comparative mitochondrial genomes of four species of Sinopodisma and phylogenetic implications (Orthoptera, Melanoplinae). Zookeys 2020, 2020, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.N.; Zhu, B.J.; Dai, L.S.; Liu, C.L. The complete mitogenome of Bombyx mori strain Dazao (Lepidoptera: Bombycidae) and comparison with other lepidopteran insects. Genomics 2013, 101, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.R.; Hsu, K.C.; Gu, S.; He, X.B.; Luo, Z.S.; Lin, H.D.; Yan, Y.R. Complete mitogenomes of four Trichiurus species: A taxonomic review of the T.lepturus species complexe. ZooKeys 2022, 1084, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nishikawa, K.; Matsui, M.; Nguyen, T.Q.; Xie, F.; Li, C.; Khatiwada, J.R.; Zhang, B.; Gong, D.; Mo, Y.; et al. Phylogenetic surveys on the newt genus Tylototriton sensu lato (Salamandridae, Caudata) reveal cryptic diversity and novel diversification promoted by historical climatic shifts. PeerJ 2018, 2018, e4384. [Google Scholar] [CrossRef]
- Rancilhac, L.; Irisarri, I.; Angelini, C.; Arntzen, J.W.; Babik, W.; Bossuyt, F.; Kunzel, S.; Luddecke, T.; Pasmans, F.; Sanchez, E.; et al. Phylotranscriptomic evidence for pervasive ancient hybridization among Old World salamanders. Mol. Phylogenet Evol 2021, 155, 106967. [Google Scholar] [CrossRef]
- Fairmaire, L. Coléoptères de l’intérieur de la Chine. Annls. Soc. Ent. Belg 1887, 31, 87–136. [Google Scholar]
- Bieñkowski, A.O.; Orlova-Bienkowskaja, M.J. Quick spread of the invasive rose flea beetle Luperomorpha Xanthodera (Fairmaire, 1888) in Europe and its first record from Russia: (Coleoptera, Chrysomelidae, Galerucinae, Alticini). Spixiana 2018, 41, 99–104. [Google Scholar]
- Del Bene, G.; Conti, B. Notes on the biology and ethology of Luperomorpha Xanthodera, a flea beetle recently introduced into Europe. Bull. Insectology 2009, 62, 61–68. [Google Scholar]
- Beenen, R.; Roques, A. Leaf and seed beetles (Coleoptera, Chrysomelidae). BioRisk 2010, 4, 267–292. [Google Scholar] [CrossRef]
- Kozlowski, M.W.; Legutowska, H. The invasive flea beetle Luperomorpha Xanthodera (Coleoptera: Chrysomelidae: Alticinae), potentially noxious to ornamental plants-first record in Poland. J. Plant Prot. Res. 2014, 54, 106–107. [Google Scholar] [CrossRef]
- Yang, X.; Siqin, G.; Nie, R.; Ruan, Y.; Li, W. Chinese Leaf Beetles; Science Press: Beijing, China, 2015; 504p. [Google Scholar]
- Fagot, J.; Libert, P. Entretiens sur les Chrysomelidae de Belgique et des régions limitrophes 6. Luperomorpha Xanthodera (Fairmaire, 1888), espèce nouvelle pour la faune belge (Chrysomelidae, Alticinae). Entomol. Faun. Entomol. 2016, 69, 81–82. [Google Scholar]
- Sady, E.A.; Kiełkiewicz, M.; Kozłowski, M.W. The rose flea beetle (Luperomorpha xanthodera, Coleoptera: Chrysomelidae), an alien species in central Poland − from an episodic occurrence in an established population. J. Plant Prot. Res. 2020, 60, 86–97. [Google Scholar] [CrossRef]
- Furth, D.G. Relationships of Palearctic and Nearctic genera of Alticinae (Coleoptera: Chrysomelidae). Entomography 1985, 3, 375–392. [Google Scholar]
- Furth, D.G.; Suzuki, K. Character correlation studies of problematic genera of Alticinae in relation to Galerucinae (Coleoptera: Chrysomelidae). Proc. Third Int. Symp. Chrysomelidae Beijing 1992, 1994, 116–135. [Google Scholar]
- Nie, R.E.; Breeschoten, T.; Timmermans, M.J.T.N.; Nadein, K.; Xue, H.J.; Bai, M.; Huang, Y.; Yang, X.K.; Vogler, A.P. The phylogeny of Galerucinae (Coleoptera: Chrysomelidae) and the performance of mitochondrial genomes in phylogenetic inference compared to nuclear rRNA genes. Cladistics 2018, 34, 113–130. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kück, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Wang, H.C.; Minh, B.Q.; Susko, E.; Roger, A.J. Modeling site heterogeneity with posterior mean site frequency profiles accelerates accurate phylogenomic estimation. Syst. Biol. 2018, 67, 216–235. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Stahlke, A.R.; Ozsoy, A.Z.; Bean, D.W.; Hohenlohe, P.A. Mitochondrial genome sequences of Diorhabda Carinata and Diorhabda Carinulata, two beetle species introduced to North America for biological control. Microbiol. Resour. Announc. 2019, 8, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Shi, Y.X.; Song, L.; Zhang, Y.; Zhang, H.F. The complete mitochondrial genome of an important agricultural pest Monolepta Hieroglyphica (Coleoptera: Chrysomelidae: Galerucinae). Mitochond. DNA Part B 2020, 5, 1820–1821. [Google Scholar] [CrossRef]
- Nie, R.E.; Wei, J.; Zhang, S.K.; Vogler, A.P.; Wu, L.; Konstantinov, A.S.; Li, W.Z.; Yang, X.K.; Xue, H.J. Diversification of mitogenomes in three sympatric Altica flea beetles (Insecta, Chrysomelidae). Zool. Scr. 2019, 48, 657–666. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. A comparative analysis of mitochondrial genomes in coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol. Biol. Evol. 2008, 25, 2499–2509. [Google Scholar] [CrossRef] [Green Version]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Boore, J.L. Complete mitochondrial genome sequence of Urechis caupo, a representative of the phylum Echiura. BMC Genom. 2004, 5, 67. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, G. The mitochondrial genomes of two walnut pests, Gastrolina Depressa Depressa and G. Depressa Thoracica (Coleoptera: Chrysomelidae), and phylogenetic analyses. PeerJ 2018, 6, e4919. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Lee, S.; Leschen, R.A.B.; Lee, S. Evolution of feeding habits of sap beetles (Coleoptera: Nitidulidae) and placement of Calonecrinae. Syst. Entomol. 2020, 45, 911–923. [Google Scholar] [CrossRef]
- Gimmel, M.L.; Bocakova, M.; Gunter, N.L.; Leschen, R. Comprehensive phylogeny of the Cleroidea (Coleoptera: Cucujiformia). Syst. Entomol. 2019, 44, 527–558. [Google Scholar] [CrossRef]
- Gillespie, J.J.; Tallamy, D.W.; Riley, E.G.; Cognato, A.I. Molecular phylogeny of rootworms and related Galerucine beetles (Coleoptera: Chrysomelidae). Zool. Scr. 2008, 37, 195–222. [Google Scholar] [CrossRef]
- Lee, C.F. The genus Paleosepharia Laboissière, 1936 in Taiwan: Review and nomenclatural changes (Coleoptera, Chrysomelidae, Galerucinae). Zookeys 2018, 19, 19–41. [Google Scholar] [CrossRef] [Green Version]
- Ge, D.; Gómez-Zurita, J.; Chesters, D.; Yang, X.; Vogler, A.P. Suprageneric systematics of flea Beetles (Chrysomelidae: Alticinae) inferred from multilocus sequence data. Mol. Phylogenet. Evol. 2012, 62, 793–805. [Google Scholar] [CrossRef]
- Gillespie, J.J.; Kjer, K.M.; Duckett, C.N.; Tallamy, D.W. Convergent evolution of cucurbitacin feeding in spatially isolated rootworm taxa (Coleoptera: Chrysomelidae; Galerucinae, Luperini). Mol. Phylogenet. Evol. 2003, 29, 161–175. [Google Scholar] [CrossRef]
- Kim, S.J.; Kjer, K.M.; Duckett, C.N. Comparison between molecular and morphological-based phylogenies of Galerucine/Alticine leaf beetles (Coleoptera: Chrysomelidae). Insect Syst. Evol. 2003, 34, 53–64. [Google Scholar] [CrossRef]
- Ge, D.; Chesters, D.; Zhang, L.; Yang, X.; Alfried, P. Anti-predator defence drives parallel morphological evolution in flea beetles. Proc. R. Soc. B Biol. Sci. 2011, 278, 2133–2141. [Google Scholar] [CrossRef] [PubMed]
- Chaboo, C.S.; Clark, S. Beetles (Coleoptera) of Peru: A survey of the families. Chrysomelidae: Galerucinae (not including Alticini). J. Kansas Entomol. Soc. 2015, 88, 361–367. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Tribe | Species | Length (bp) | G + C% | A + T% | AT-Skew | GC-Skew | GenBank No. |
|---|---|---|---|---|---|---|---|---|
| Galerucinae | Alticini | Agasicles hygrophila | 15,917 | 24.9 | 75.2 | 0.05 | −0.19 | NC_028332 |
| Altica cirsicola | 15,864 | 22.2 | 77.8 | 0.03 | −0.21 | NC_042876 | ||
| Altica fragariae | 16,220 | 22.0 | 78 | 0.03 | −0.22 | NC_042875 | ||
| Altica viridicyanea | 16,706 | 22.3 | 77.8 | 0.04 | −0.23 | NC_048472 | ||
| Argopistes tsekooni | 16,552 | 20.5 | 79.5 | 0.04 | −0.19 | NC_045929 | ||
| Chaetocnema pelagica | 16,331 | 23.1 | 76.9 | 0.06 | −0.20 | NC_041170 | ||
| Macrohaltica subplicata | 15,840 | 22.0 | 78 | 0.02 | −0.22 | NC_041169 | ||
| Phyllotreta striolata | 15,689 | 24.2 | 75.8 | 0.05 | −0.26 | NC_045901 | ||
| Psylliodes chlorophana | 14,561 | 23.7 | 76.3 | 0.05 | −0.22 | NC_053362 | ||
| Luperomorpha hainana | 16,081 | 22.2 | 77.8 | 0.05 | −0.25 | MF960124 | ||
| Luperomorpha xanthodera | 16,021 | 20 | 80 | 0.03 | −0.16 | ON631248 | ||
| Incertae sedis | Paleosepharia posticata | 15,729 | 20.2 | 79.8 | 0.02 | −0.17 | NC_033532 | |
| Podagricomela nigricollis | 16,756 | 21.3 | 78.8 | 0.05 | −0.18 | NC_041423 | ||
| Galerucini | Diorhabda carinata | 16,232 | 22.1 | 77.9 | 0.03 | −0.19 | NC_042945 | |
| Diorhabda carinulata | 16,298 | 21 | 78.9 | 0.03 | −0.18 | NC_042946 | ||
| Galeruca daurica | 16,615 | 21.8 | 78.1 | 0.04 | −0.20 | NC_027114 | ||
| Ophraella communa | 16,553 | 22.6 | 77.4 | 0.06 | −0.18 | NC_039710 | ||
| Luperini | Aulacophora indica | 16,246 | 21.4 | 78.6 | 0.03 | −0.20 | NC_047467 | |
| Aulacophora lewisii | 15,691 | 21.1 | 78.9 | 0.03 | −0.18 | NC_039712 | ||
| Diabrotica barberi | 16,366 | 20.8 | 79.2 | 0.04 | −0.18 | NC_022935 | ||
| Hoplasoma unicolor | 14,568 | 22.2 | 77.8 | 0.05 | −0.23 | NC_041168 | ||
| Monolepta hieroglyphica | 15,963 | 19.7 | 80.3 | 0.02 | −0.18 | NC_057489 | ||
| Monolepta occifluvis | 15,998 | 20.7 | 79.2 | 0.03 | −0.20 | NC_045838 | ||
| Monolepta quadriguttata | 16,130 | 19.7 | 80.3 | 0.02 | −0.19 | NC_039711 | ||
| Oidini | Oides livida | 16,127 | 20.0 | 79.9 | 0.05 | −0.14 | MF960098 | |
| Chrysomelinae | Chrysomelini | Chrysomela vigintipunctata | 17,474 | 21.7 | 78.2 | 0.04 | −0.21 | NC_050933 |
| Chrysolina aeruginosa | 16,335 | 24.5 | 75.5 | 0.07 | −0.25 | NC_052915 |
| Gene | Direction | Location (bp) | Size (bp) | Codon | Anti-Codon | IGN | ||
|---|---|---|---|---|---|---|---|---|
| from | to | Start | Stop | |||||
| tRNAIle | F | 1 | 62 | 62 | • | • | GAT | |
| tRNAGln | R | 63 | 131 | 69 | • | • | TTG | 0 |
| tRNAMet | F | 131 | 199 | 69 | • | • | CAT | -1 |
| ND2 | F | 200 | 1210 | 1011 | ATT | TAA | • | 0 |
| tRNATrp | F | 1211 | 1274 | 64 | • | • | TCA | 0 |
| tRNACys | R | 1267 | 1328 | 62 | • | • | GCA | -8 |
| tRNATyr | R | 1329 | 1391 | 63 | • | • | GTA | 0 |
| COⅠ | F | 1384 | 2926 | 1543 | ATT | T | • | -8 |
| tRNALeu | F | 2927 | 2991 | 65 | • | • | TAA | 0 |
| COⅡ | F | 2992 | 3676 | 685 | ATG | T | • | 0 |
| tRNALys | F | 3677 | 3746 | 70 | • | • | TTT | 0 |
| tRNAAsp | F | 3747 | 3809 | 63 | • | • | GTC | 0 |
| ATP8 | F | 3810 | 3965 | 156 | ATC | TAA | • | 0 |
| ATP6 | F | 3959 | 4633 | 675 | ATG | TAA | • | -7 |
| COⅢ | F | 4633 | 5421 | 789 | ATG | TAA | • | -1 |
| tRNAGly | F | 5424 | 5488 | 65 | • | • | TCC | 2 |
| ND3 | F | 5489 | 5842 | 354 | ATT | TAG | • | 0 |
| tRNAAla | F | 5841 | 5904 | 64 | • | • | TGC | -2 |
| tRNAArg | F | 5905 | 5968 | 64 | • | • | TCG | 0 |
| tRNAAsn | F | 5966 | 6033 | 68 | • | • | GTT | -3 |
| tRNASer | F | 6034 | 6100 | 67 | • | • | TCT | 0 |
| tRNAGlu | F | 6102 | 6165 | 64 | • | • | TTC | 1 |
| tRNAPhe | R | 6165 | 6227 | 63 | • | • | GAA | -1 |
| ND5 | R | 6228 | 7932 | 1705 | ATT | T | • | 0 |
| tRNAHis | R | 7933 | 7995 | 63 | • | • | GTG | 0 |
| ND4 | R | 8005 | 9325 | 1321 | ATG | T | • | 9 |
| ND4L | R | 9319 | 9600 | 282 | ATG | TAA | • | -7 |
| tRNAThr | F | 9603 | 9665 | 63 | • | • | TGT | 2 |
| tRNAPro | R | 9666 | 9729 | 64 | • | • | TGG | 0 |
| ND6 | F | 9732 | 10,235 | 504 | ATT | TAA | • | 2 |
| CYTB | F | 10,235 | 11,374 | 1140 | ATG | TAG | • | -1 |
| tRNASer | F | 11,373 | 11,439 | 67 | • | • | TGA | -2 |
| ND1 | R | 11,457 | 12,407 | 951 | TTG | TAG | • | 17 |
| tRNALeu | R | 12,409 | 12,473 | 65 | • | • | TAG | 1 |
| 16S rRNA | R | 12,474 | 13,753 | 1280 | • | • | • | 0 |
| tRNAVal | R | 13,754 | 13,821 | 68 | • | • | TAC | 0 |
| 12S rRNA | R | 13,822 | 14,633 | 812 | • | • | • | 0 |
| A + T rich region | F | 14,634 | 16,021 | 1388 | • | • | • | 0 |
| Genes | Size (bp) | Nucleotides Composition | ATskew | GCskew | |||||
|---|---|---|---|---|---|---|---|---|---|
| A (%) | T (%) | G (%) | C (%) | A + T (%) | G + C (%) | ||||
| Complete mitogenome | 16,021 | 41.3 | 38.7 | 8.4 | 11.6 | 80 | 20 | 0.032 | −0.160 |
| PCGs | 11,125 | 33.9 | 44.3 | 11.1 | 10.7 | 78.2 | 21.8 | −0.133 | 0.018 |
| 1st codon position | 3711 | 35.0 | 38.2 | 16.7 | 10.1 | 73.2 | 26.8 | −0.043 | 0.247 |
| 2nd codon position | 3707 | 21.1 | 47.9 | 13.4 | 17.5 | 69.0 | 29 | −0.388 | −0.135 |
| 3rd codon position | 3707 | 45.5 | 46.7 | 3.2 | 4.6 | 92.2 | 7.8 | −0.014 | −0.181 |
| tRNA | 1432 | 41.9 | 38.8 | 8.7 | 10.6 | 80.7 | 19.3 | 0.038 | −0.098 |
| rRNA | 2092 | 44.6 | 39.6 | 5.5 | 10.3 | 84.2 | 15.8 | 0.059 | −0.304 |
| 16S rRNA | 1280 | 45.1 | 38.7 | 5.7 | 10.5 | 83.8 | 16.2 | 0.076 | −0.296 |
| 12S rRNA | 812 | 43.8 | 41.0 | 5.2 | 10.0 | 84.8 | 15.2 | 0.033 | −0.316 |
| A + T rich region | 1388 | 44.1 | 43.2 | 5.0 | 7.7 | 87.3 | 12.7 | 0.010 | −0.213 |
| Amino Acid | Codon | Count | RSCU | Amino Acid | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|
| Phe | UUU | 325 | 1.86 | Tyr | UAU | 162 | 1.79 |
| UUC | 24 | 0.14 | UAC | 19 | 0.21 | ||
| Leu2 | UUA | 524 | 5.22 | His | CAU | 59 | 1.66 |
| UUG | 21 | 0.21 | CAC | 12 | 0.34 | ||
| Leu1 | CUU | 22 | 0.22 | Gln | CAA | 57 | 1.84 |
| CUC | 3 | 0.03 | CAG | 5 | 0.16 | ||
| CUA | 27 | 0.27 | Asn | AAU | 190 | 1.83 | |
| CUG | 5 | 0.05 | AAC | 18 | 0.17 | ||
| Ile | AUU | 391 | 1.87 | Lys | AAA | 102 | 1.87 |
| AUC | 27 | 0.13 | AAG | 7 | 0.13 | ||
| Met | AUA | 241 | 1.85 | Asp | GAU | 62 | 1.8 |
| AUG | 20 | 0.15 | GAC | 7 | 0.13 | ||
| Val | GUU | 66 | 1.8 | Glu | GAA | 69 | 1.86 |
| GUC | 5 | 0.14 | GAG | 5 | 0.14 | ||
| GUA | 69 | 1.88 | Cys | UGU | 30 | 1.88 | |
| GUG | 7 | 0.19 | UGC | 2 | 0.13 | ||
| Ser2 | UCU | 107 | 2.58 | Trp | UGA | 88 | 1.91 |
| UCC | 7 | 0.17 | UGG | 4 | 0.09 | ||
| UCA | 88 | 2.12 | Arg | CGU | 19 | 1.36 | |
| UCG | 3 | 0.07 | CGC | 1 | 0.07 | ||
| Pro | CCU | 66 | 2.06 | CGA | 34 | 2.43 | |
| CCC | 8 | 0.25 | CGG | 2 | 0.14 | ||
| CCA | 53 | 1.66 | Ser1 | AGU | 27 | 0.56 | |
| CCG | 1 | 0.03 | AGC | 3 | 0.07 | ||
| Thr | ACU | 90 | 2.03 | AGA | 85 | 2.05 | |
| ACC | 11 | 0.25 | AGG | 12 | 0.29 | ||
| ACA | 76 | 1.72 | Gly | GGU | 50 | 1.05 | |
| ACG | 0 | 0 | GGC | 6 | 0.13 | ||
| Ala | GCU | 63 | 1.81 | GGA | 110 | 2.3 | |
| GCC | 16 | 0.46 | GGG | 25 | 0.52 | ||
| GCA | 59 | 1.7 | * | UAA | 0 | 0 | |
| GCG | 1 | 0.03 | UAG | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Yan, B.; He, H.; Xu, X.; Ruan, Y.; Yang, M. Characterization of the Complete Mitochondrial Genome of a Flea Beetle Luperomorpha xanthodera (Coleoptera: Chrysomelidae: Galerucinae) and Phylogenetic Analysis. Genes 2023, 14, 414. https://doi.org/10.3390/genes14020414
Li J, Yan B, He H, Xu X, Ruan Y, Yang M. Characterization of the Complete Mitochondrial Genome of a Flea Beetle Luperomorpha xanthodera (Coleoptera: Chrysomelidae: Galerucinae) and Phylogenetic Analysis. Genes. 2023; 14(2):414. https://doi.org/10.3390/genes14020414
Chicago/Turabian StyleLi, Jingjing, Bin Yan, Hongli He, Xiaoli Xu, Yongying Ruan, and Maofa Yang. 2023. "Characterization of the Complete Mitochondrial Genome of a Flea Beetle Luperomorpha xanthodera (Coleoptera: Chrysomelidae: Galerucinae) and Phylogenetic Analysis" Genes 14, no. 2: 414. https://doi.org/10.3390/genes14020414
APA StyleLi, J., Yan, B., He, H., Xu, X., Ruan, Y., & Yang, M. (2023). Characterization of the Complete Mitochondrial Genome of a Flea Beetle Luperomorpha xanthodera (Coleoptera: Chrysomelidae: Galerucinae) and Phylogenetic Analysis. Genes, 14(2), 414. https://doi.org/10.3390/genes14020414