
Prenatal Clinical Findings in RASA1-Related Capillary Malformation-Arteriovenous Malformation Syndrome

 , , , , ,
, , , , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
3. Patients and Results
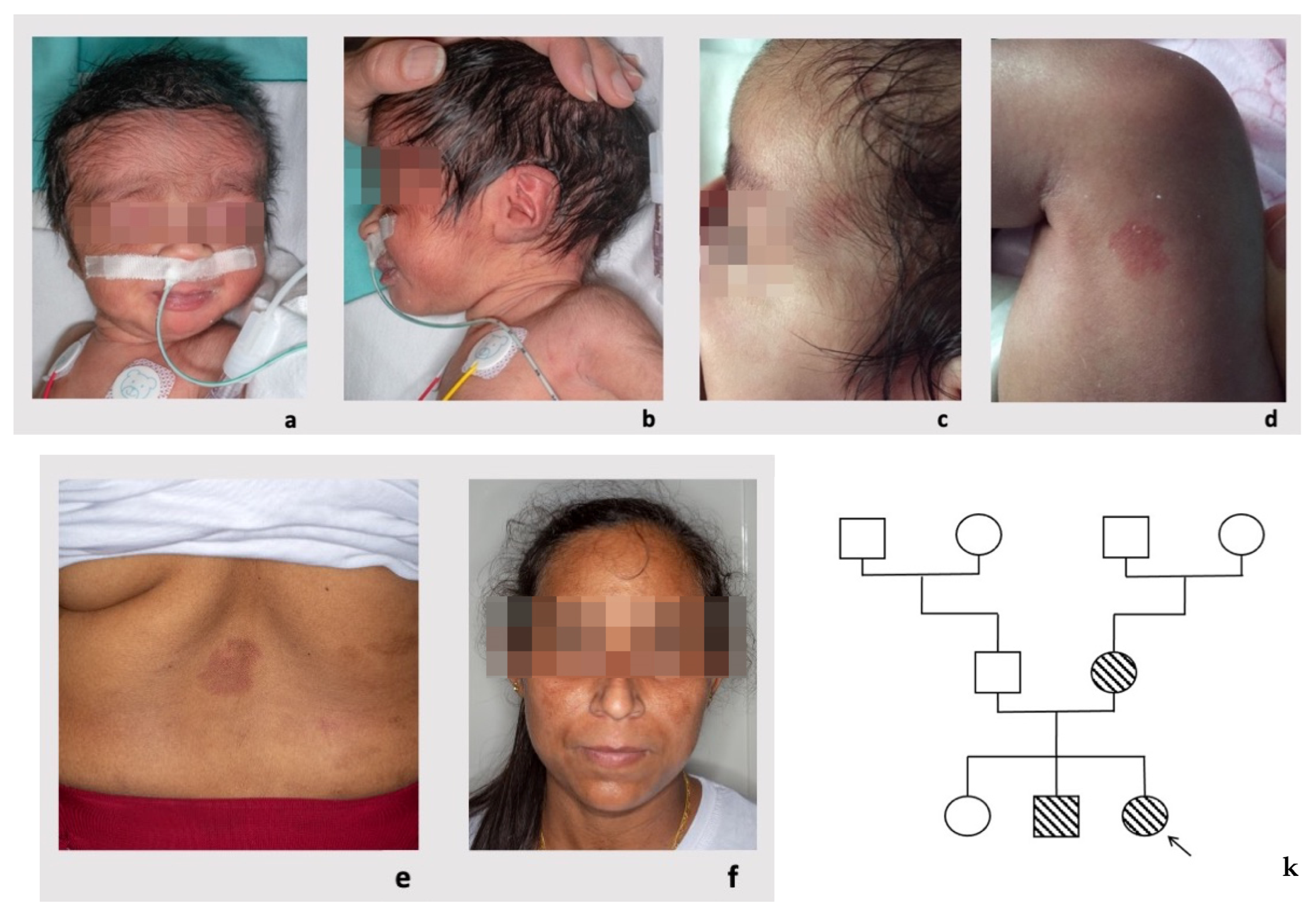
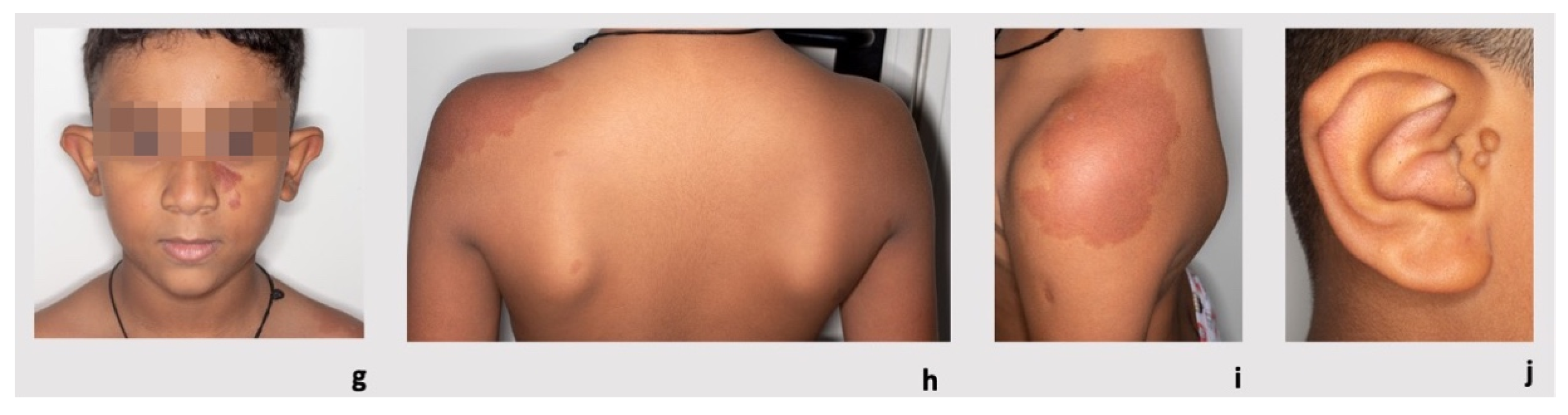
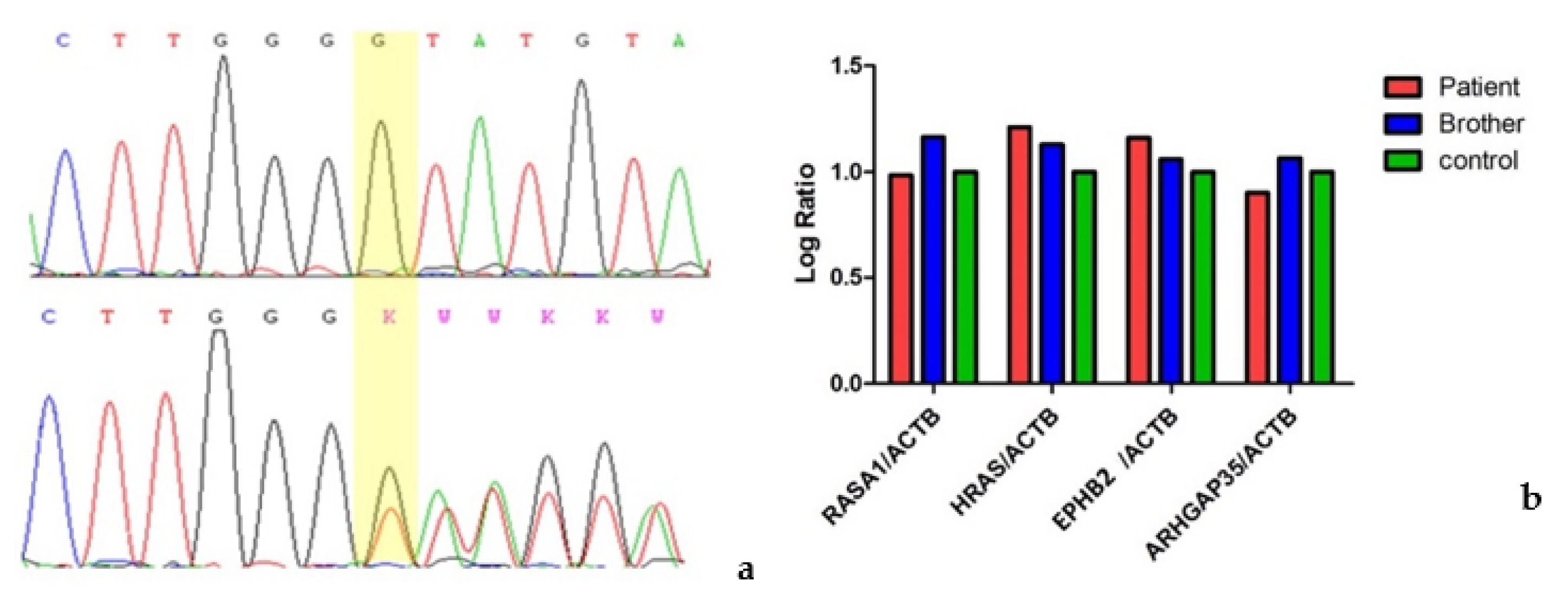
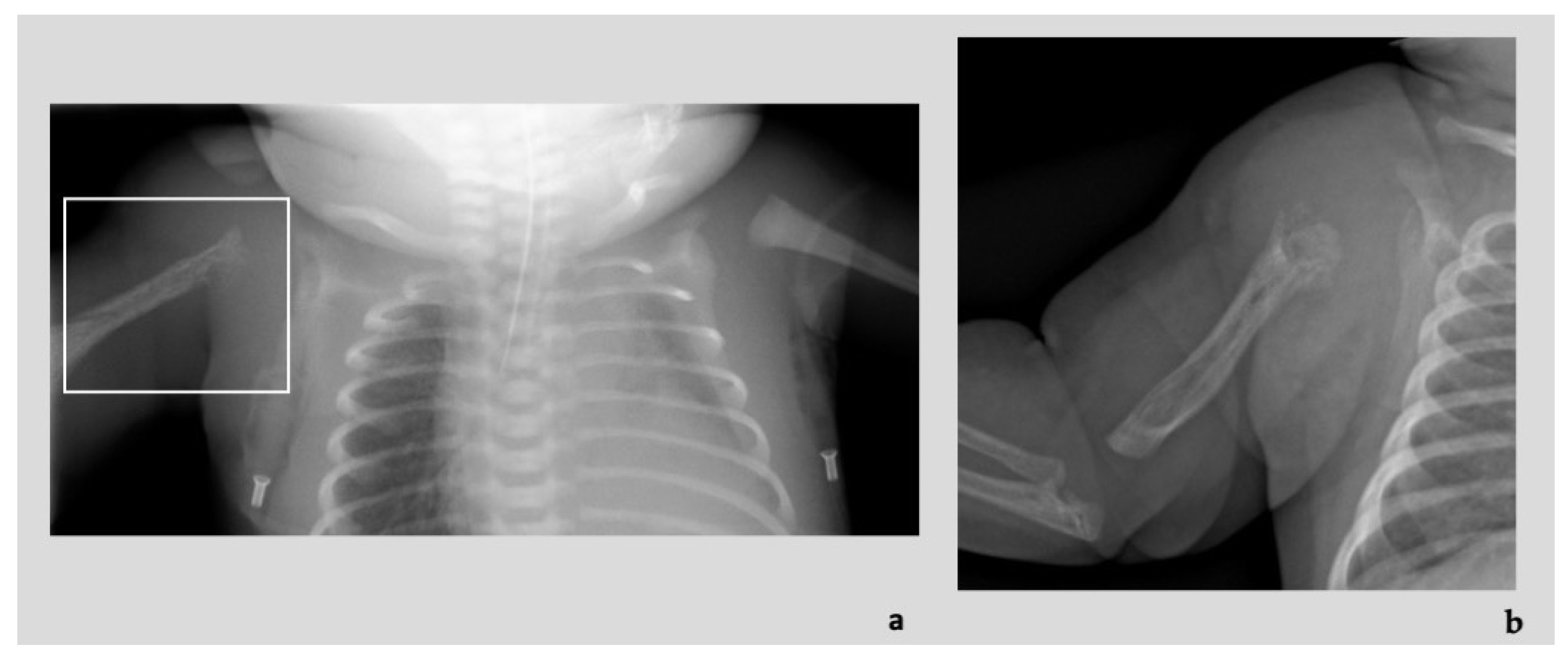
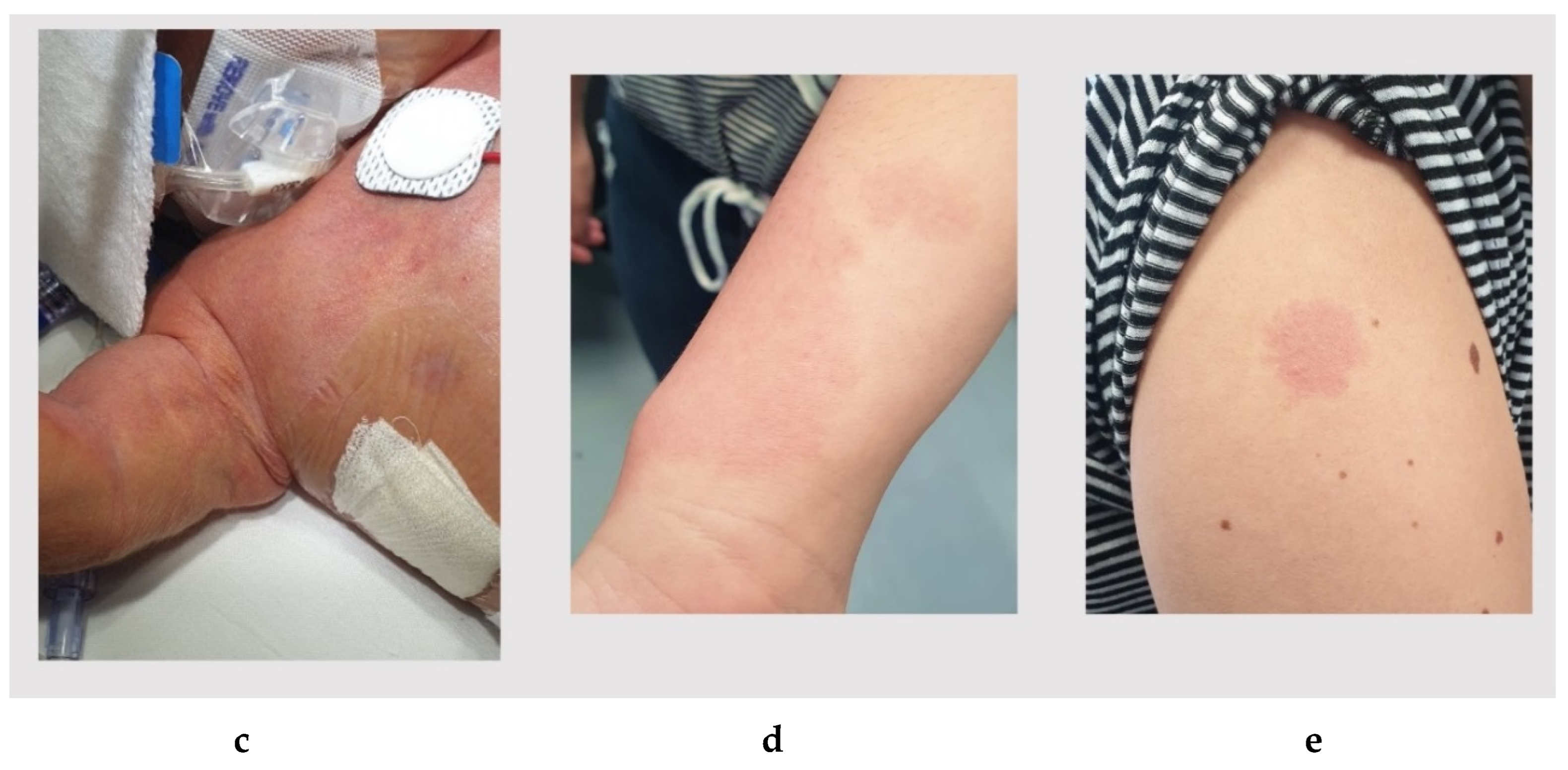
3.1. Patient 1
3.2. Patient 2
3.3. Patient 3
3.4. Patient 4
3.5. Review of the Literature
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eerola, I.; Boon, L.M.; Mulliken, J.B.; Burrows, P.E.; Dompmartin, A.; Watanabe, S.; Vanwijck, R.; Vikkula, M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am. J. Hum. Genet. 2003, 73, 1240–1249. [Google Scholar] [CrossRef] [Green Version]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmuller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Lubeck, B.A.; Lapinski, P.E.; Bauler, T.J.; Oliver, J.A.; Hughes, E.D.; Saunders, T.L.; King, P.D. Blood vascular abnormalities in Rasa1(R780Q) knockin mice: Implications for the pathogenesis of capillary malformation-arteriovenous malformation. Am. J. Pathol. 2014, 184, 3163–3169. [Google Scholar] [CrossRef] [Green Version]
- King, P.D.; Lubeck, B.A.; Lapinski, P.E. Nonredundant functions for Ras GTPase-activating proteins in tissue homeostasis. Sci Signal. 2013, 6, re1. [Google Scholar] [CrossRef] [Green Version]
- Lapinski, P.E.; Lubeck, B.A.; Chen, D.; Doosti, A.; Zawieja, S.D.; Davis, M.J.; King, P.D. RASA1 regulates the function of lymphatic vessel valves in mice. J. Clin. Investig. 2017, 127, 2569–2585. [Google Scholar] [CrossRef] [Green Version]
- Bayrak-Toydemir, P.; Stevenson, D.A. Capillary Malformation-Arteriovenous Malformation Syndrome. In GeneReviews® [Internet]. Seattle (WA); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Boccara, O.; Mazereeuw, J.; Martin, L.; Bessis, D.; Hubiche, T.; Chiaverini, C.; Dompmartin, A.; Mallet, S.; Miquel, J.; Aubert, H.; et al. Groupe de Recherche Clinique de Dermatologie Pédiatrique (GRDP); Filière Maladies rares Dermatologiques (FIMARAD). Central nervous system screening in capillary malformation-arteriovenous malformation syndrome: An observational study. J. Am. Acad. Dermatol. 2022, 87, 914–916. [Google Scholar] [CrossRef]
- Weitz, N.A.; Lauren, C.T.; Behr, G.G.; Wu, J.K.; Kandel, J.J.; Meyers, P.M.; Sultan, S.; Anyane-Yeboa, K.; Morel, K.D.; Garzon, M.C. Clinical spectrum of capillary malformation-arteriovenous malformation syndrome presenting to a pediatric dermatology practice: A retrospective study. Pediatr. Dermatol. 2015, 32, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Henkemeyer, M.; Rossi, D.J.; Holmyard, D.P.; Puri, M.C.; Mbamalu, G.; Harpal, K.; Shih, T.S.; Jacks, T.; Pawson, T. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 1995, 377, 695–701. [Google Scholar] [CrossRef]
- Jafry, M.; Sidbury, R. RASopathies. Clin. Dermatol. 2020, 38, 455–461. [Google Scholar] [CrossRef]
- Mangels, R.; Blumenfeld, Y.J.; Homeyer, M.; Mrazek-Pugh, B.; Hintz, S.R.; Hudgins, L. RASopathies: A significant cause of polyhydramnios? Prenat. Diagn. 2021, 41, 362–367. [Google Scholar] [CrossRef]
- Zenker, M. Clinical overview on RASopathies. Am. J. Med. Genet. C Semin. Med. Genet. 2022, 190, 414–424. [Google Scholar] [CrossRef]
- Rauen, K.A. Defining RASopathy. Dis. Model Mech. 2022, 15, dmm049344. [Google Scholar] [CrossRef]
- Revencu, N.; Boon, L.M.; Mendola, A.; Cordisco, M.R.; Dubois, J.; Clapuyt, P.; Hammer, F.; Amor, D.J.; Irvine, A.D.; Baselga, E.; et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum. Mutat. 2013, 34, 1632–1641. [Google Scholar] [CrossRef]
- Durrington, H.J.; Firth, H.V.; Patient, C.; Belham, M.; Jayne, D.; Burrows, N.; Morrell, N.W.; Chilvers, E.R. A novel RASA1 mutation causing capillary malformation-arteriovenous malformation (CM-AVM) presenting during pregnancy. Am. J. Med. Genet. A. 2013, 161, 1690–1694. [Google Scholar] [CrossRef]
- Kwong, Y.; Cartmill, M.; Jaspan, T.; Suri, M. Fetal MRI demonstrating vein of Galen malformations in two successive pregnancies—A previously unreported occurrence. Childs Nerv. Syst. 2015, 31, 1033–1035. [Google Scholar] [CrossRef]
- Overcash, R.T.; Gibu, C.K.; Jones, M.C.; Ramos, G.A.; Andreasen, T.S. Maternal and fetal capillary malformation–arteriovenous malformation (CM–AVM) due to a novel RASA1 mutation presenting with prenatal nonimmune hydrops fetalis. Am. J. Med. Genet. A 2015, 167, 2440–2443. [Google Scholar] [CrossRef] [PubMed]
- Grillner, P.; Söderman, M.; Holmin, S.; Rodesch, G. A spectrum of intracranial vascular high-flow arteriovenous shunts in RASA1 mutations. Childs Nerv. Syst. 2016, 32, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Macmurdo, C.F.; Wooderchak-Donahue, W.; Bayrak-Toydemir, P.; Le, J.; Wallenstein, M.B.; Milla, C.; Teng, J.M.C.; Bernstein, J.A.; Stevenson, D.A. RASA1 somatic mutation and variable expressivity in capillary malformation/arteriovenous malformation (CM/AVM) syndrome. Am. J. Med. Genet. A 2016, 170, 1450–1454. [Google Scholar] [CrossRef]
- Saliou, G.; Eyries, M.; Iacobucci, M.; Knebel, J.F.; Waill, M.C.; Coulet, F.; Ozanne, A.; Soubrier, F. Clinical and genetic findings in children with central nervous system arteriovenous fistulas. Ann. Neurol. 2017, 82, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Lacalm, A.; Fichez, A.; Broussin, B.; Abel, C.; Lacombe, D.; Guibaud, L. Prenatal diagnosis of cerebral and extracerebral high-flow lesions revealing familial capillary malformation-arteriovenous malformation (CM-AVM) syndrome. Ultrasound Obstet. Gynecol. 2018, 51, 409–411. [Google Scholar] [CrossRef] [Green Version]
- Wooderchak-Donahue, W.L.; Johnson, P.; McDonald, J.; Blei, F.; Berenstein, A.; Sorscher, M.; Mayer, J.; Scheuerle, A.E.; Lewis, T.; Grimmer, J.F.; et al. Expanding the clinical and molecular findings in RASA1 capillary malformation-arteriovenous malformation. Eur. J. Hum. Genet. 2018, 26, 1521–1536. [Google Scholar] [CrossRef] [Green Version]
- D’Amours, G.; Brunel-Guitton, C.; Delrue, M.-A.; Dubois, J.; Laberge, S.; Soucy, J.-F. Prenatal pleural effusions and chylothorax: An unusual presentation for CM-AVM syndrome due to RASA1. Am. J. Med. Genet. A 2020, 182, 2454–2460. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, P.; Holder, S.E.; Carton, J.; Wakelin, S. The protean manifestations of RASA1 gene mutation. Clin. Exp. Dermatol. 2019, 44, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Gallipoli, A.; MacLean, G.G.; Walia, J.S.; Sehgal, A. Congenital Chylothorax and Hydrops Fetalis: A Novel Neonatal Presentation of RASA1 Mutation. Pediatrics 2021, 147, e2020011601. [Google Scholar] [CrossRef]
- Guimaraes, M.J.; Gomes, J.; Lopes, G.; Caldas, R.; Brito, C. Capillary malformation-arteriovenous malformation syndrome associated with basilar artery aneurysm. Pediatr. Dermatol. 2022, 39, 662–663. [Google Scholar] [CrossRef]
- Westenius, E.; Sahlin, E.; Conner, P.; Lindstrand, A.; Iwarsson, E. Diagnostic yield using whole-genome sequencing and an in-silico gene panel of 281 genes associated with non-immune hydrops fetalis in a clinical setting. Ultrasound Obstet. Gynecol. 2022, 60, 487–493. [Google Scholar] [CrossRef] [PubMed]
- OMIM—Online Mendelian Inheritance in Man®. Available online: https://www.omim.org/ (accessed on 9 December 2022).
- ExAC and gnomAD. Available online: http://gnomad.broadinstitute.org/ (accessed on 12 December 2022).
- Varsome. Available online: https://varsome.com/ (accessed on 12 December 2022).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar (accessed on 12 December 2022).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Human Splicing Finder 3.1. Available online: http://umd.be/Redirect.html (accessed on 12 December 2022).
- Croonen, E.A.; Nillesen, W.M.; Stuurman, K.E.; Oudesluijs, G.; Van de Laar, I.M.B.M.; Martens, L.; Ockeloen, C.; Mathijssen, I.B.; Schepens, M.; Ruiterkamp-Versteeg, M.; et al. Prenatal diagnostic testing of the Noonan syndrome genes in fetuses with abnormal ultrasound findings. Eur. J. Hum. Genet. 2013, 21, 936–942. [Google Scholar] [CrossRef] [Green Version]
- Myers, A.; Bernstein, J.A.; Brennan, M.-L.; Curry, C.; Esplin, E.D.; Fisher, J.; Homeyer, M.; Manning, M.A.; Muller, E.A.; Niemi, A.-K.; et al. Perinatal features of the RASopathies: Noonan syndrome, cardiofaciocutaneous syndrome and Costello syndrome. Am. J. Med. Genet. A. 2014, 164, 2814–2821. [Google Scholar]
- Bakker, M.; Pajkrt, E.; Mathijssen, I.B.; Bilardo, C.M. Targeted ultrasound examination and DNA testing for Noonan syndrome, in fetuses with increased nuchal translucency and normal karyotype. Prenat. Diagn. 2011, 31, 833–840. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Feature | Literature Review Frequency (%) | This Study P1 | This Study P2 | This Study P3 | This Study P4 | Total |
|---|---|---|---|---|---|---|
| [11,14,15,16,17,18,19,20,21,22,23,24,25,26,27] | ||||||
| Capillary malformations, postnatal findings (skin) | 11/15 (73.3%) | + | + | + | + | 15/19 (78.9%) |
| Increased fetal nuchal thickness | 2/3 (66.7%) | - | - | + | n/r | 3/6 (50%) |
| Vascular malformations, including postnatal findings (AVMs and AVFs) | 13/20 (65%) | + | - | - | + | 15/24 (62.5%) |
| Polyhydramnios | 8/21 (38.1%) | + | + | + | - | 11/25 (44%) |
| Deceased | 6/20 (30%) | - | - | - | - | 6/24 (25%) |
| Cardiac failure | 5/20 (25%) | + | - | - | + | 7/24 (29.2%) |
| Pleural effusion | 5/21 (23.8%) | + | - | - | + | 7/25 (28%) |
| Non-immune hydrops fetalis | 5/21 (23.8%) | + | - | - | - | 6/25 (24%) |
| Structural cardiac anomalies | 4/21 (19%) | - | - | + (PFO) | + (CM, PFO) | 6/25 (24%) |
| Chylothorax | 3/20 (15%) | + | - | - | - | 4/24 (16.7%) |
| Parkes Weber Syndrome | 2/16 (12.5%) | - | - | - | + | 3/20 (15%) |
| Ascites | 2/20 (10%) | - | - | - | + | 3/24 (12.5%) |
| In utero drainage/shunt | 2/20 (10%) | + | + | - | - | 4/24 (16.7%) |
| VGAM | 2/20 (10%) | - | - | - | - | 2/24 (8.3%) |
| Renal anomalies | 2/21 (9.5%) | - | - | - | - | 2/25 (8%) |
| Pericardial effusion | 1/20 (5%) | + | - | - | - | 2/24 (8.3%) |
| Basilar artery aneurism | 1/20 (5%) | - | - | - | - | 1/24 (4.2%) |
| Literature Review Frequency (%) [11,14,15,16,17,18,19,20,21,22,23,24,25,26,27] | This Study P1 | This Study P2 | This Study P3 | This Study P4 | Total | |
|---|---|---|---|---|---|---|
| Inheritance | ||||||
| De novo RASA1 variant | 4/17 (23.5%) | - | - | - | - | 4/21 (19%) |
| Inherited RASA1 variant | 13/17 (76.5%) | + (mother) | + (mother) | + (mother) | + (mother) | 17/21 (81%) |
| Variant information | ||||||
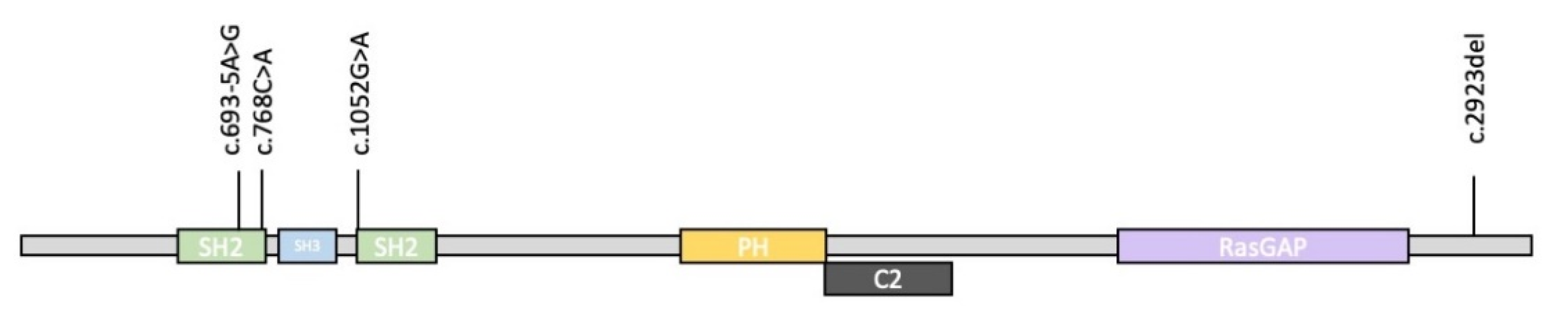
| Nonsense variant | 7/18 (39%) | - | - | + c.1052G>A | + c.768C>A | 9/22 (41%) |
| Frameshift variant | 7/18 (39%) | + c.2923del | - | - | - | 8/22 (36%) |
| Splicing variant | 2/18 (11%) | - | + c.693-5A>G | - | - | 3/22 (14%) |
| Missense variant | 1/18 (5.5%) | - | - | - | - | 1/22 (4.5%) |
| Deletion | 1/18 (5.5%) | - | - | - | - | 1/22 (4.5%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coccia, E.; Valeri, L.; Zuntini, R.; Caraffi, S.G.; Peluso, F.; Pagliai, L.; Vezzani, A.; Pietrangiolillo, Z.; Leo, F.; Melli, N.; et al. Prenatal Clinical Findings in RASA1-Related Capillary Malformation-Arteriovenous Malformation Syndrome. Genes 2023, 14, 549. https://doi.org/10.3390/genes14030549
Coccia E, Valeri L, Zuntini R, Caraffi SG, Peluso F, Pagliai L, Vezzani A, Pietrangiolillo Z, Leo F, Melli N, et al. Prenatal Clinical Findings in RASA1-Related Capillary Malformation-Arteriovenous Malformation Syndrome. Genes. 2023; 14(3):549. https://doi.org/10.3390/genes14030549
Chicago/Turabian StyleCoccia, Emanuele, Lara Valeri, Roberta Zuntini, Stefano Giuseppe Caraffi, Francesca Peluso, Luca Pagliai, Antonietta Vezzani, Zaira Pietrangiolillo, Francesco Leo, Nives Melli, and et al. 2023. "Prenatal Clinical Findings in RASA1-Related Capillary Malformation-Arteriovenous Malformation Syndrome" Genes 14, no. 3: 549. https://doi.org/10.3390/genes14030549
APA StyleCoccia, E., Valeri, L., Zuntini, R., Caraffi, S. G., Peluso, F., Pagliai, L., Vezzani, A., Pietrangiolillo, Z., Leo, F., Melli, N., Fiorini, V., Greco, A., Lepri, F. R., Pisaneschi, E., Marozza, A., Carli, D., Mussa, A., Radio, F. C., Conti, B., ... Garavelli, L. (2023). Prenatal Clinical Findings in RASA1-Related Capillary Malformation-Arteriovenous Malformation Syndrome. Genes, 14(3), 549. https://doi.org/10.3390/genes14030549