
Human Genes Involved in the Interaction between Host and Gut Microbiome: Regulation and Pathogenic Mechanisms

 ,
, 
 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
3. Results
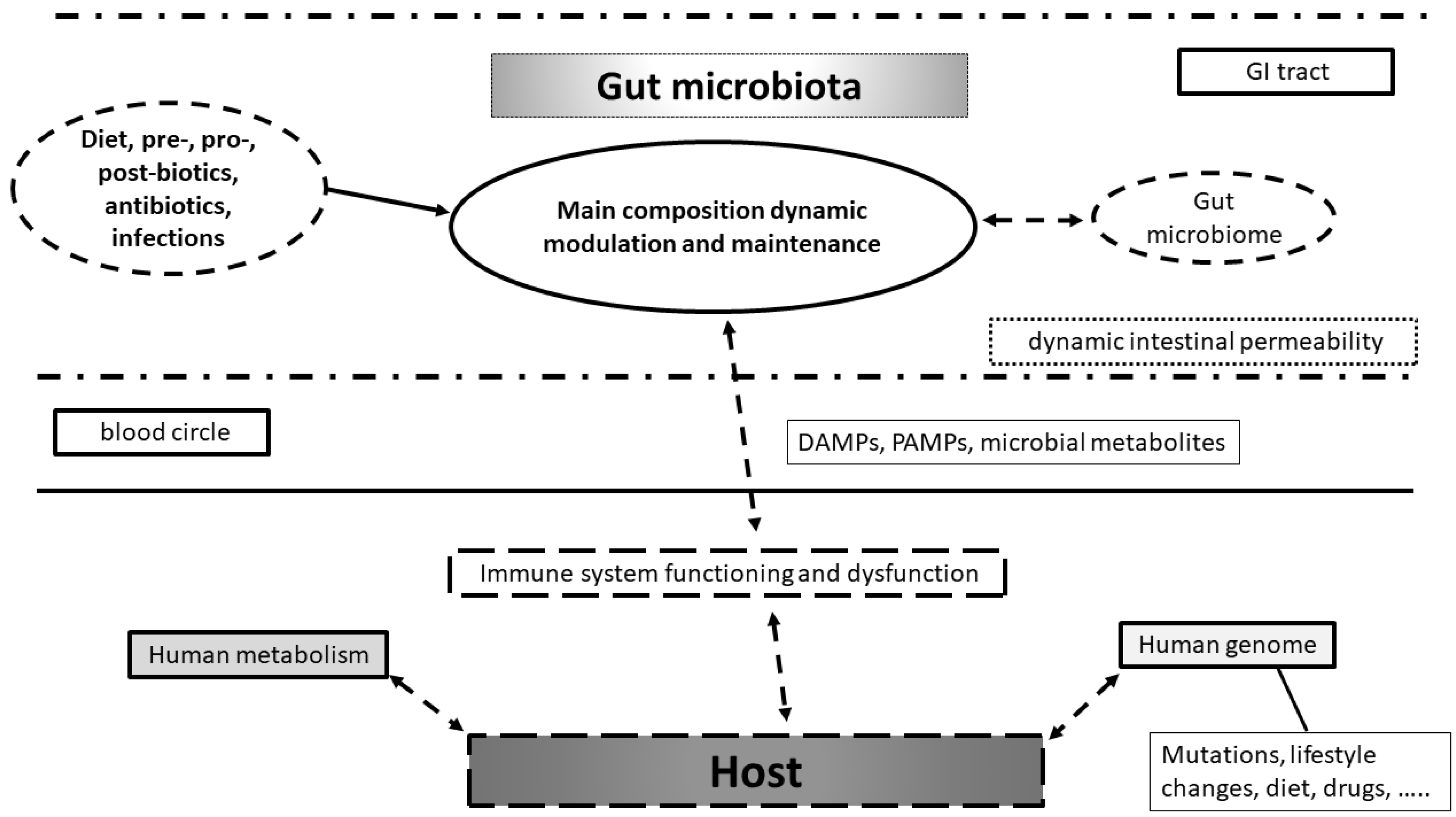
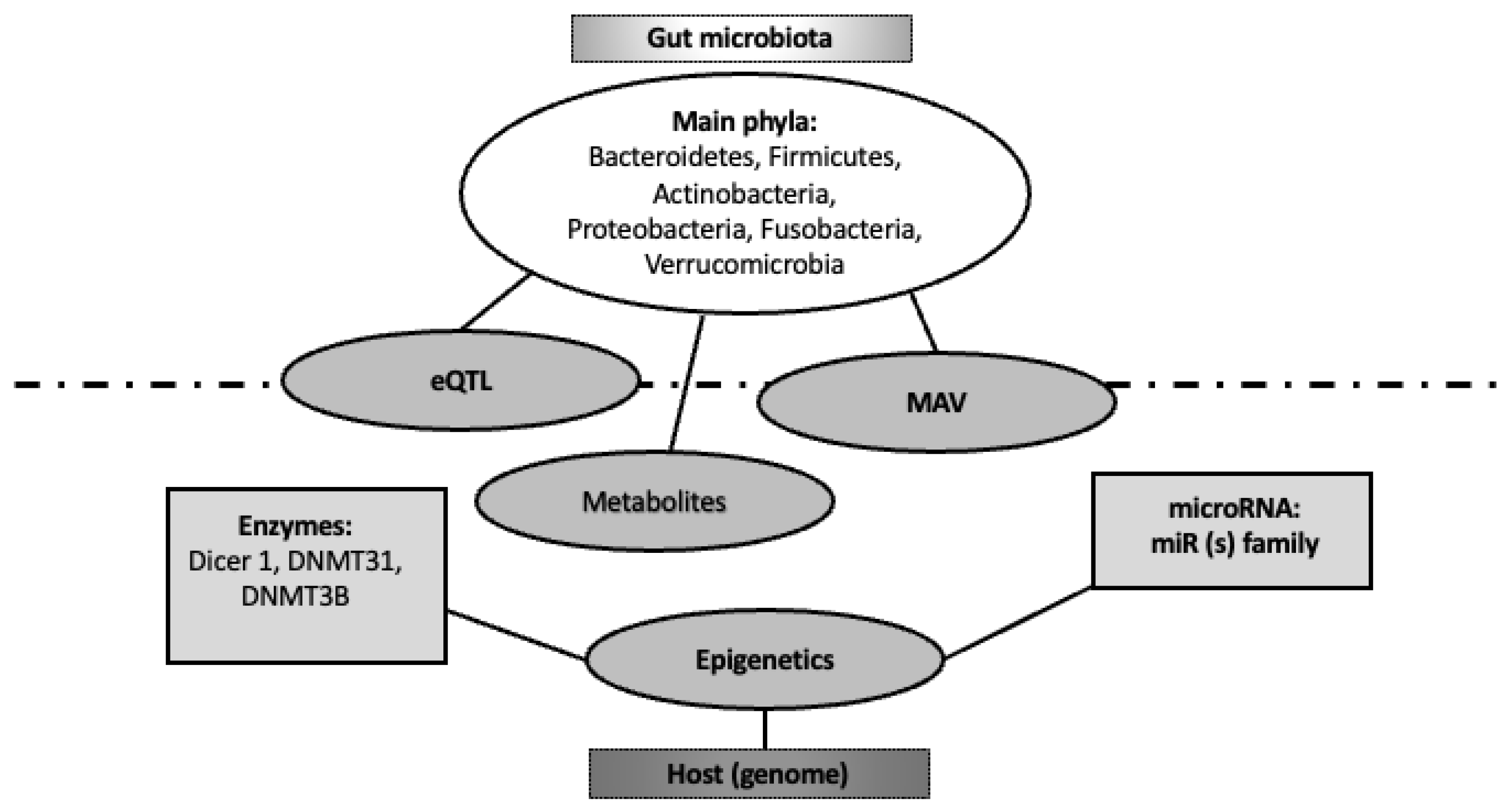
3.1. Human Genomic Control by the Microbiome
3.2. Genomic Control of Gut Microbiome by the Host
4. Gut Metagenome in Health and Disease
5. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Scarpellini, E.; Basilico, M.; Rinninella, E.; Carbone, F.; Schol, J.; Rasetti, C.; Abenavoli, L.; Santori, P. Probiotics and gut health. Minerva Gastroenterol. 2021, 67, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Künstner, A.; Schilf, P.; Busch, H.; Ibrahim, S.M.; Hirose, M. Changes of Gut Microbiota by Natural mtDNA Variant Differences Augment Susceptibility to Metabolic Disease and Ageing. Int. J. Mol. Sci. 2022, 23, 1056. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Attaye, I.; Warmbrunn, M.V.; Boot, A.N.; van der Wolk, S.C.; Hutten, B.A.; Daams, J.G.; Herrema, H.; Nieuwdorp, M. A Systematic Review and Meta-analysis of Dietary Interventions Modulating Gut Microbiota and Cardiometabolic Diseases—Striving for New Standards in Microbiome Studies. Gastroenterology 2022, 162, 1911–1932. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Saris, W.H.; Tarnopolsky, M.A. Controlling food intake and energy balance: Which macronutrient should we select? Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.; Garelli, S.; Rampelli, S.; Agostini, A.; Matysik, S.; D’Amico, F.; Krautbauer, S.; Mazza, R.; Salituro, N.; Fanelli, F.; et al. Multi-omics gut microbiome signatures in obese women: Role of diet and uncontrolled eating behavior. BMC Med. 2022, 20, 500. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Amit, I.; Regev, A.; Hacohen, N. Strategies to discover regulatory circuits of the mammalian immune system. Nat. Rev. Immunol. 2011, 11, 873–880. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Elinav, E. Metagenomic cross-talk: The regulatory interplay between immunogenomics and the microbiome. Genome Med. 2015, 7, 120. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [PubMed]
- Shivaji, S. We are not alone: A case for the human microbiome in extra intestinal diseases. Gut Pathog. 2017, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Neveu, V.; Nicolas, G.; Amara, A.; Salek, R.M.; Scalbert, A. The human microbial exposome: Expanding the Exposome-Explorer database with gut microbial metabolites. Sci. Rep. 2023, 13, 1946. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, J.; Wang, H.; Lu, W.; Lee, Y.K.; Zhao, J.; Zhang, H. Machine learning framework for gut microbiome biomarkers discovery and modulation analysis in large-scale obese population. BMC Genom. 2022, 23, 850. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Bautista Balbás, L.A.; Sandino Gómez, R.; Gil Conesa, M.; Velasco Guijarro, O.; Rodríguez Caravaca, G.; Jou Rivera, F.; Navasquillo Lorda, M.Á.; Martín Carmena, E. Seroprevalence of SARS-CoV2 Infections in Health Care Personnel in a Long-Term Care Institution After the First Wave of the Pandemic: A Cross-Sectional Study. Workplace Health Saf. 2023, 21650799221135587. [Google Scholar] [CrossRef]
- Heinken, A.; Hertel, J.; Acharya, G.; Ravcheev, D.A.; Nyga, M.; Okpala, O.E.; Hogan, M.; Magnúsdóttir, S.; Martinelli, F.; Nap, B.; et al. Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Diener, C.; Dai, C.L.; Wilmanski, T.; Baloni, P.; Smith, B.; Rappaport, N.; Hood, L.; Magis, A.T.; Gibbons, S.M. Genome–microbiome interplay provides insight into the determinants of the human blood metabolome. Nat. Metab. 2022, 4, 1560–1572. [Google Scholar] [CrossRef] [PubMed]
- Stappenbeck, T.S.; Hooper, L.V.; Manchester, J.K.; Wong, M.H.; Gordon, J.I. Laser capture microdissection of mouse intestine: Characterizing mRNA and protein expression; and profiling intermediary metabolism in specified cell populations. Methods Enzym. 2002, 356, 167–196. [Google Scholar]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014, 516, 94–98. [Google Scholar] [CrossRef]
- Larsson, E.; Tremaroli, V.; Lee, Y.S.; Koren, O.; Nookaew, I.; Fricker, A.; Nielsen, J.; Ley, R.; Bäckhed, F. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut 2012, 61, 1124–1131. [Google Scholar] [CrossRef]
- Sommer, F.; Nookaew, I.; Sommer, N.; Fogelstrand, P.; Bäckhed, F. Site-specific programming of the host epithelial transcriptome by the gut microbiota. Genome Biol. 2015, 16, 62. [Google Scholar] [CrossRef]
- Dasgupta, S.; Kasper, D.L. Traffic control at the “Gut-GALT crossroads”. Cell Res. 2013, 23, 590–591. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Kong, Y.; Kleinstein, S.H.; Subramanian, S.; Ahern, P.P.; Gordon, J.I.; Medzhitov, R. Analysis of gene–environment interactions in postnatal development of the mammalian intestine. Proc. Natl. Acad. Sci. USA 2015, 112, 1929–1936. [Google Scholar] [CrossRef]
- Rawls, J.F.; Mahowald, M.A.; Ley, R.E.; Gordon, J.I. Reciprocal Gut Microbiota Transplants from Zebrafish and Mice to Germ-free Recipients Reveal Host Habitat Selection. Cell 2006, 127, 423–433. [Google Scholar] [CrossRef]
- Yadav, M.; Chauhan, N.S. Role of gut-microbiota in disease severity and clinical outcomes. Brief. Funct. Genom. 2022, elac037. [Google Scholar] [CrossRef]
- Lam, T.J.; Ye, Y. Meta-analysis of microbiome association networks reveal patterns of dysbiosis in diseased microbiomes. Sci. Rep. 2022, 12, 17482. [Google Scholar] [CrossRef]
- Leone, V.; Gibbons, S.M.; Martinez, K.; Hutchison, A.L.; Huang, E.Y.; Cham, C.M.; Pierre, J.F.; Heneghan, A.F.; Nadimpalli, A.; Hubert, N.; et al. Effects of Diurnal Variation of Gut Microbes and High-Fat Feeding on Host Circadian Clock Function and Metabolism. Cell Host Microbe 2015, 17, 681–689. [Google Scholar] [CrossRef]
- Takahashi, K.; Sugi, Y.; Nakano, K.; Tsuda, M.; Kurihara, K.; Hosono, A.; Kaminogawa, S. Epigenetic Control of the Host Gene by Commensal Bacteria in Large Intestinal Epithelial Cells. J. Biol. Chem. 2011, 286, 35755–35762. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, T.; Osborne, L.C.; Saenz, S.A.; Kobuley, D.; Ziegler, C.G.K.; Mullican, S.E.; Choi, I.; Grunberg, S.; Sinha, R.; Wynosky-Dolfi, M.; et al. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature 2013, 504, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Frank, C.L.; Lickwar, C.R.; Guturu, H.; Rube, T.; Wenger, A.M.; Chen, J.; Bejerano, G.; Crawford, G.E.; Rawls, J.F. Microbiota modulate transcription in the intestinal epithelium without remodeling the accessible chromatin landscape. Genome Res. 2014, 24, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Obata, T.; Kunisawa, J.; Sato, S.; Ivanov, I.I.; Lamichhane, A.; Takeyama, N.; Kamioka, M.; Sakamoto, M.; Matsuki, T.; et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 2014, 345, 1254009. [Google Scholar] [CrossRef]
- Zhang, Q.; Pan, Y.; Yan, R.; Zeng, B.; Wang, H.; Zhang, X.; Li, W.; Wei, H.; Liu, Z. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat. Immunol. 2015, 16, 918–926. [Google Scholar] [CrossRef]
- El Aidy, S.; van Baarlen, P.; Derrien, M.; Lindenbergh-Kortleve, D.J.; Hooiveld, G.; Levenez, F.; Doré, J.; Dekker, J.; Samsom, J.N.; Nieuwenhuis, E.E.S.; et al. Temporal and spatial interplay of microbiota and intestinal mucosa drive establishment of immune homeostasis in conventionalized mice. Mucosal Immunol. 2012, 5, 567–579. [Google Scholar] [CrossRef]
- Ganal, S.C.; Sanos, S.L.; Kallfass, C.; Oberle, K.; Johner, C.; Kirschning, C.; Lienenklaus, S.; Weiss, S.; Staeheli, P.; Aichele, P.; et al. Priming of Natural Killer Cells by Nonmucosal Mononuclear Phagocytes Requires Instructive Signals from Commensal Microbiota. Immunity 2012, 37, 171–186. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; DeRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Olszak, T.; An, D.; Zeissig, S.; Vera, M.P.; Richter, J.; Franke, A.; Glickman, J.N.; Siebert, R.; Baron, R.M.; Kasper, D.L.; et al. Microbial Exposure During Early Life Has Persistent Effects on Natural Killer T Cell Function. Science 2012, 336, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Ley, R. The Human Gut Microbiome: Ecology and Recent Evolutionary Changes. Annu. Rev. Microbiol. 2011, 65, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M., Jr.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef]
- Tims, S.; Derom, C.; Jonkers, D.M.; Vlietinck, R.; Saris, W.H.; Kleerebezem, M.; De Vos, W.M.; Zoetendal, E.G. Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J. 2013, 7, 707–717. [Google Scholar] [CrossRef]
- Simões, C.D.; Maukonen, J.; Kaprio, J.; Rissanen, A.; Pietiläinen, K.H.; Saarela, M. Habitual Dietary Intake Is Associated with Stool Microbiota Composition in Monozygotic Twins. J. Nutr. 2013, 143, 417–423. [Google Scholar] [CrossRef]
- Hansen, E.E.; Lozupone, C.A.; Rey, F.E.; Wu, M.; Guruge, J.L.; Narra, A.; Goodfellow, J.; Zaneveld, J.R.; McDonald, D.T.; Goodrich, J.A.; et al. Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proc. Natl. Acad. Sci. USA 2011, 108, 4599–4606. [Google Scholar] [CrossRef]
- Williams, R.W.; Gu, J.; Qi, S.; Lu, L. The genetic structure of recombinant inbred mice: High-resolution consensus maps for complex trait analysis. Genome Biol. 2001, 2, RESEARCH0046. [Google Scholar] [CrossRef]
- McKnite, A.M.; Perez-Munoz, M.E.; Lu, L.; Williams, E.G.; Brewer, S.; Andreux, P.A.; Bastiaansen, J.W.M.; Wang, X.; Kachman, S.D.; Auwerx, J.; et al. Murine Gut Microbiota Is Defined by Host Genetics and Modulates Variation of Metabolic Traits. PLoS ONE 2012, 7, e39191. [Google Scholar] [CrossRef]
- Blekhman, R.; Goodrich, J.K.; Huang, K.; Sun, Q.; Bukowski, R.; Bell, J.T.; Spector, T.D.; Keinan, A.; Ley, R.E.; Gevers, D.; et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015, 16, 191. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.A.; Frank, D.N.; Pace, N.R.; Gordon, J.I. Metagenomic Approaches for Defining the Pathogenesis of Inflammatory Bowel Diseases. Cell Host Microbe 2008, 3, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Fiocchi, C. Genes and ‘in-vironment’: How will our concepts on the pathophysiology of inflammatory bowel disease develop in the future? Dig. Dis. 2012, 30, 2–11. [Google Scholar] [CrossRef]
- Frank, D.N.; Robertson, C.; Hamm, C.M.; Kpadeh, Z.; Zhang, T.; Chen, H.; Zhu, W.; Sartor, R.B.; Boedeker, E.C.; Harpaz, N.; et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm. Bowel Dis. 2011, 17, 179–184. [Google Scholar] [CrossRef]
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Schwab, M.; Schäffeler, E.; Schlee, M.; Herrlinger, K.R.; Stallmach, A.; Noack, F.; Fritz, P.; et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 2004, 53, 1658–1664. [Google Scholar] [CrossRef]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef]
- Markowitz, R.H.G.; LaBella, A.L.; Shi, M.; Rokas, A.; Capra, J.A.; Ferguson, J.F.; Mosley, J.D.; Bordenstein, S.R. Microbiome-associated human genetic variants impact phenome-wide disease risk. Proc. Natl. Acad. Sci. USA 2022, 119, e2200551119. [Google Scholar] [CrossRef]
- Wang, J.; Thingholm, L.B.; Skiecevičienė, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.-A.; Rühlemann, M.C.; Szymczak, S.; et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef]
- Kolde, R.; Franzosa, E.A.; Rahnavard, G.; Hall, A.B.; Vlamakis, H.; Stevens, C.; Daly, M.J.; Xavier, R.J.; Huttenhower, C. Host genetic variation and its microbiome interactions within the Human Microbiome Project. Genome Med. 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Flynn, E.D.; Tsu, A.L.; Kasela, S.; Kim-Hellmuth, S.; Aguet, F.; Ardlie, K.G.; Bussemaker, H.J.; Mohammadi, P.; Lappalainen, T. Transcription factor regulation of eQTL activity across individuals and tissues. PLoS Genet. 2022, 18, e1009719. [Google Scholar] [CrossRef]
- Muehlbauer, A.L.; Richards, A.L.; Alazizi, A.; Burns, M.B.; Gomez, A.; Clayton, J.B.; Petrzelkova, K.; Cascardo, C.; Resztak, J.; Wen, X.; et al. Interspecies variation in hominid gut microbiota controls host gene regulation. Cell Rep. 2021, 37, 110057. [Google Scholar] [CrossRef]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Clavel, T.; Lepage, P.; Charrier, C. “The Family Coriobacteriaceae” in The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, F., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 201–238. [Google Scholar]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef]
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Zhu, F.; Hart, J.; Roalstad, S.; Graves, J.; Lynch, S.; Waubant, E.; US Network of Pediatric MS Centers. Gut microbiota in early pediatric multiple sclerosis: A case−control study. Eur. J. Neurol. 2016, 23, 1308–1321. [Google Scholar] [CrossRef]
- Leonard, M.M.; Valitutti, F.; Karathia, H.; Pujolassos, M.; Kenyon, V.; Fanelli, B.; Troisi, J.; Subramanian, P.; Camhi, S.; Colucci, A.; et al. Microbiome signatures of progression toward celiac disease onset in at-risk children in a longitudinal prospective cohort study. Proc. Natl. Acad. Sci. USA 2021, 118, e2020322118. [Google Scholar] [CrossRef]
- Leiva-Gea, I.; Sánchez-Alcoholado, L.; Martín-Tejedor, B.; Castellano-Castillo, D.; Moreno-Indias, I.; Urda-Cardona, A.; Tinahones, F.J.; Fernández-García, J.C.; Queipo-Ortuño, M.I. Gut Microbiota Differs in Composition and Functionality between Children with Type 1 Diabetes and MODY2 and Healthy Control Subjects: A Case-Control Study. Diabetes Care 2018, 41, 2385–2395. [Google Scholar] [CrossRef]
- Groot, H.E.; Sierra, L.E.V.; Said, M.A.; Lipsic, E.; Karper, J.C.; van der Harst, P. Genetically Determined ABO Blood Group and its Associations with Health and Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 830–838. [Google Scholar] [CrossRef]
- Rühlemann, M.C.; Hermes, B.M.; Bang, C.; Doms, S.; Moitinho-Silva, L.; Thingholm, L.B.; Frost, F.; Degenhardt, F.; Wittig, M.; Kässens, J.; et al. Genome-wide association study in 8,956 German individuals identifies influence of ABO histo-blood groups on gut microbiome. Nat. Genet. 2021, 53, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed]
- Kummen, M.; Mayerhofer, C.C.K.; Vestad, B.; Broch, K.; Awoyemi, A.; Storm-Larsen, C.; Ueland, T.; Yndestad, A.; Hov, J.R.; Trøseid, M. Gut Microbiota Signature in Heart Failure Defined from Profiling of 2 Independent Cohorts. J. Am. Coll. Cardiol. 2018, 71, 1184–1186. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Yang, G.; Chen, Y.; Zhao, X.; Qian, H.; Liu, Y.; Chen, S.; Shi, G. Characteristics of the Urinary Microbiome from Patients with Gout: A Prospective Study. Front. Endocrinol. 2020, 11, 272. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Greenblum, S.; Turnbaugh, P.J.; Borenstein, E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2012, 109, 594–599. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Okuno, Y.; Tsujimoto, G. MicroRNA: Biogenetic and Functional Mechanisms and Involvements in Cell Differentiation and Cancer. J. Pharmacol. Sci. 2006, 101, 267–270. [Google Scholar] [CrossRef]
- Ohland, C.L.; Macnaughton, W.K. Probiotic bacteria and intestinal epithelial barrier function. Am. J. Physiol. Liver Physiol. 2010, 298, G807–G819. [Google Scholar] [CrossRef]
- Lee, H.-J. Microbe-Host Communication by Small RNAs in Extracellular Vesicles: Vehicles for Transkingdom RNA Transportation. Int. J. Mol. Sci. 2019, 20, 1487. [Google Scholar] [CrossRef] [PubMed]
- Assmann, T.; Cuevas-Sierra, A.; Riezu-Boj, J.; Milagro, F.; Martínez, J. Comprehensive Analysis Reveals Novel Interactions between Circulating MicroRNAs and Gut Microbiota Composition in Human Obesity. Int. J. Mol. Sci. 2020, 21, 9509. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| MAV | Gut Microbial Association | Associated Pathologic Expression |
|---|---|---|
| rs3749147 | Eggerthella | Gout |
| rs11751024 | Lachnospiraceae (CAG-882) | Type 1 diabetes |
| rs11751024 | Lachnospiraceae (CAG-882) | Celiac disease |
|
rs11751024
rs9357092 | Lachnospiraceae (CAG-882) Coriobacteriaceae (f) | Multiple sclerosis |
| rs3758348 | Faecalibacterium Bifidobacterium bifidum | Deep vein thrombosis, chronic pulmonary heart failure |
| rs11751024 | Lachnospiraceae (CAG-882) | Psoriasis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boccuto, L.; Tack, J.; Ianiro, G.; Abenavoli, L.; Scarpellini, E. Human Genes Involved in the Interaction between Host and Gut Microbiome: Regulation and Pathogenic Mechanisms. Genes 2023, 14, 857. https://doi.org/10.3390/genes14040857
Boccuto L, Tack J, Ianiro G, Abenavoli L, Scarpellini E. Human Genes Involved in the Interaction between Host and Gut Microbiome: Regulation and Pathogenic Mechanisms. Genes. 2023; 14(4):857. https://doi.org/10.3390/genes14040857
Chicago/Turabian StyleBoccuto, Luigi, Jan Tack, Gianluca Ianiro, Ludovico Abenavoli, and Emidio Scarpellini. 2023. "Human Genes Involved in the Interaction between Host and Gut Microbiome: Regulation and Pathogenic Mechanisms" Genes 14, no. 4: 857. https://doi.org/10.3390/genes14040857
APA StyleBoccuto, L., Tack, J., Ianiro, G., Abenavoli, L., & Scarpellini, E. (2023). Human Genes Involved in the Interaction between Host and Gut Microbiome: Regulation and Pathogenic Mechanisms. Genes, 14(4), 857. https://doi.org/10.3390/genes14040857