
An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review

Abstract
:1. Introduction
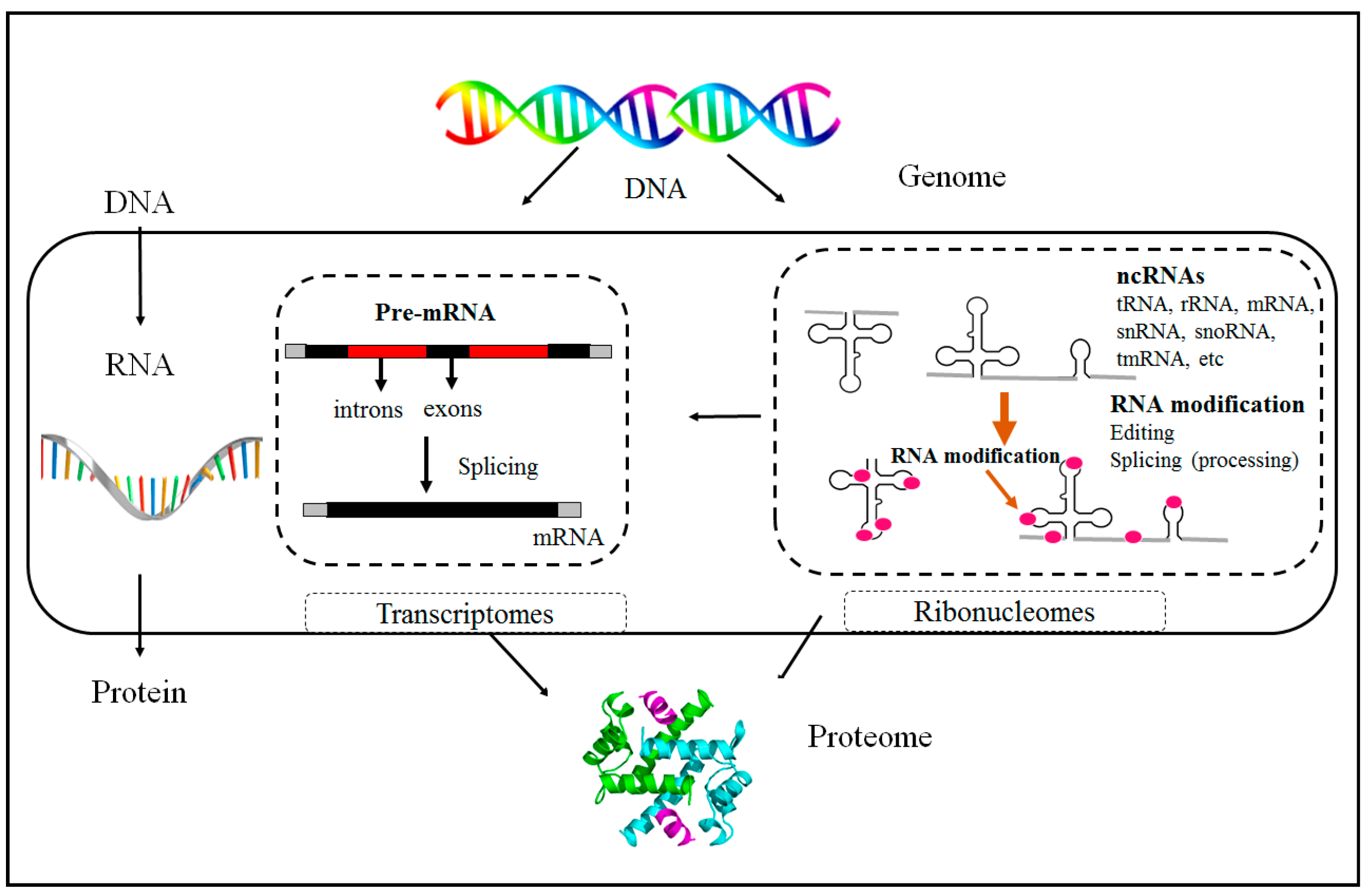
1.1. Epigenetics Overview
1.2. Non-Coding RNAs
1.2.1. N6-Methyladenosine
1.2.2. m6A Regulates miRNA Processing
1.2.3. 7-Methylguanosine (m7G)
1.2.4. 2-O-Methylation (2-O-Me)
1.3. Environmental Epigenetic Influences
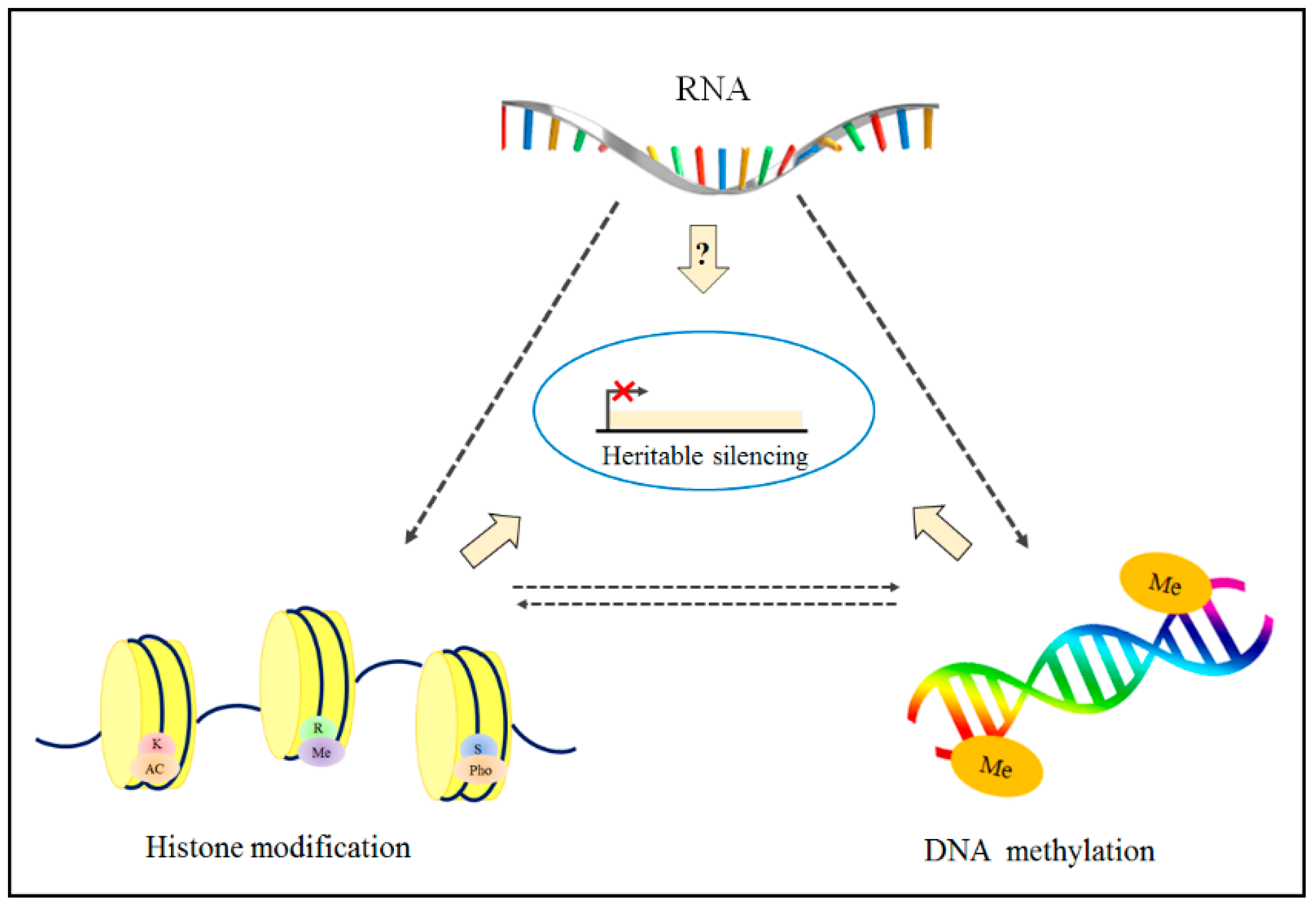
1.4. Epigenetic Modifications on Chromatin
1.5. Application of Epigenetics in Personalized Medicine
2. Epigenetic Abnormalities in Human Cancer and Brain Diseases
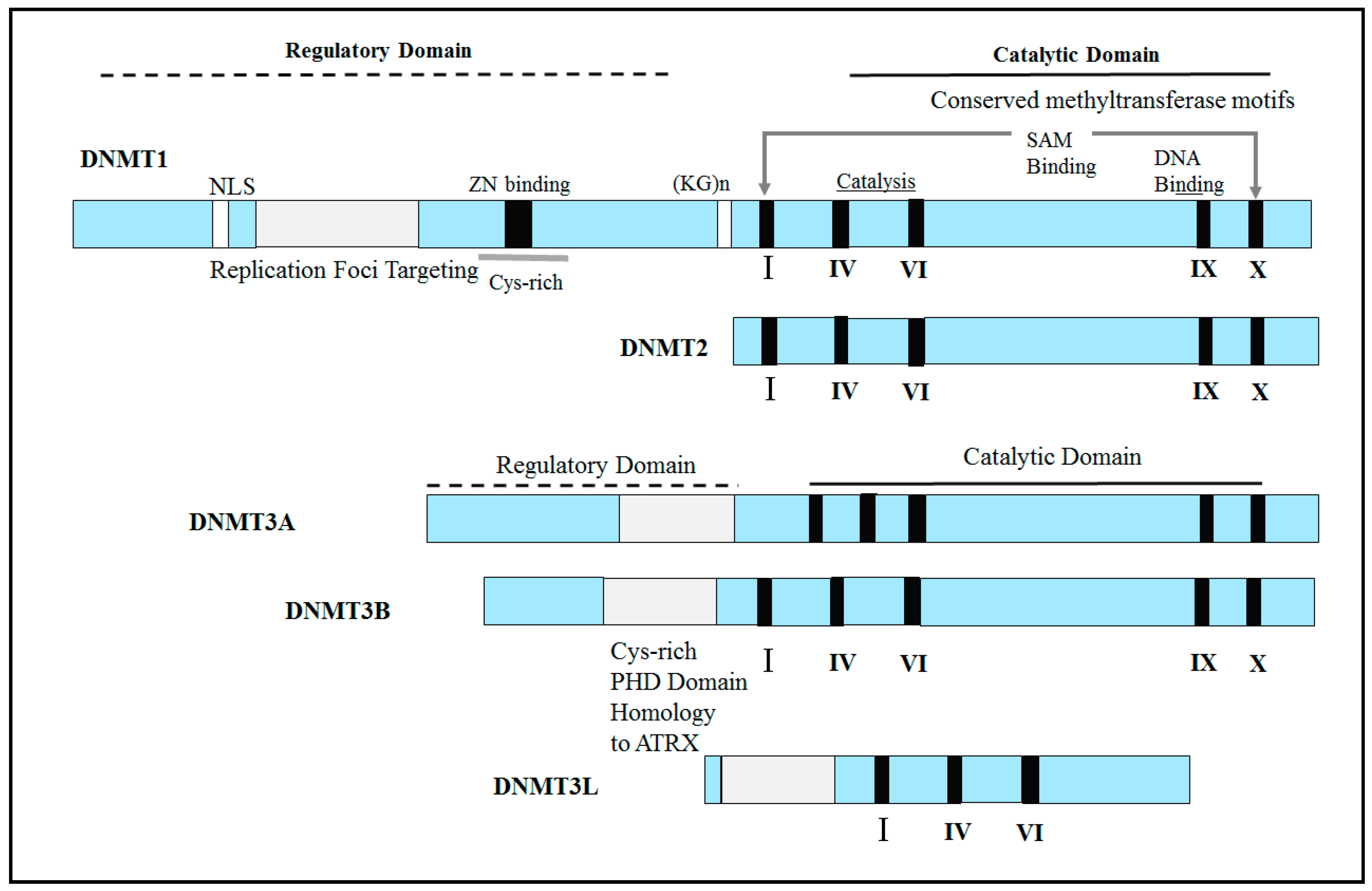
2.1. Abnormal Expression of DNA Methyltransferases (DNMTs) in Human Cancers
2.2. Relationship between DNA Methylation and Histone Modifications
2.3. Histone Modifiers in Cancer
2.4. Epigenetics and Brain Diseases
2.4.1. Bipolar and Schizophrenia Disorders
2.4.2. HSAN1 Disorder
2.4.3. Immunodeficiency, Centromeric Region Instability, and Facial Anomalies (ICF) Syndrome
2.4.4. Rett Syndrome
2.4.5. ATR-X Syndrome
2.4.6. Cornelia de Lange Syndrome
2.4.7. Rubinstein–Taybi Syndrome
2.4.8. Coffin–Lowry Syndrome
2.4.9. Kabuki Syndrome
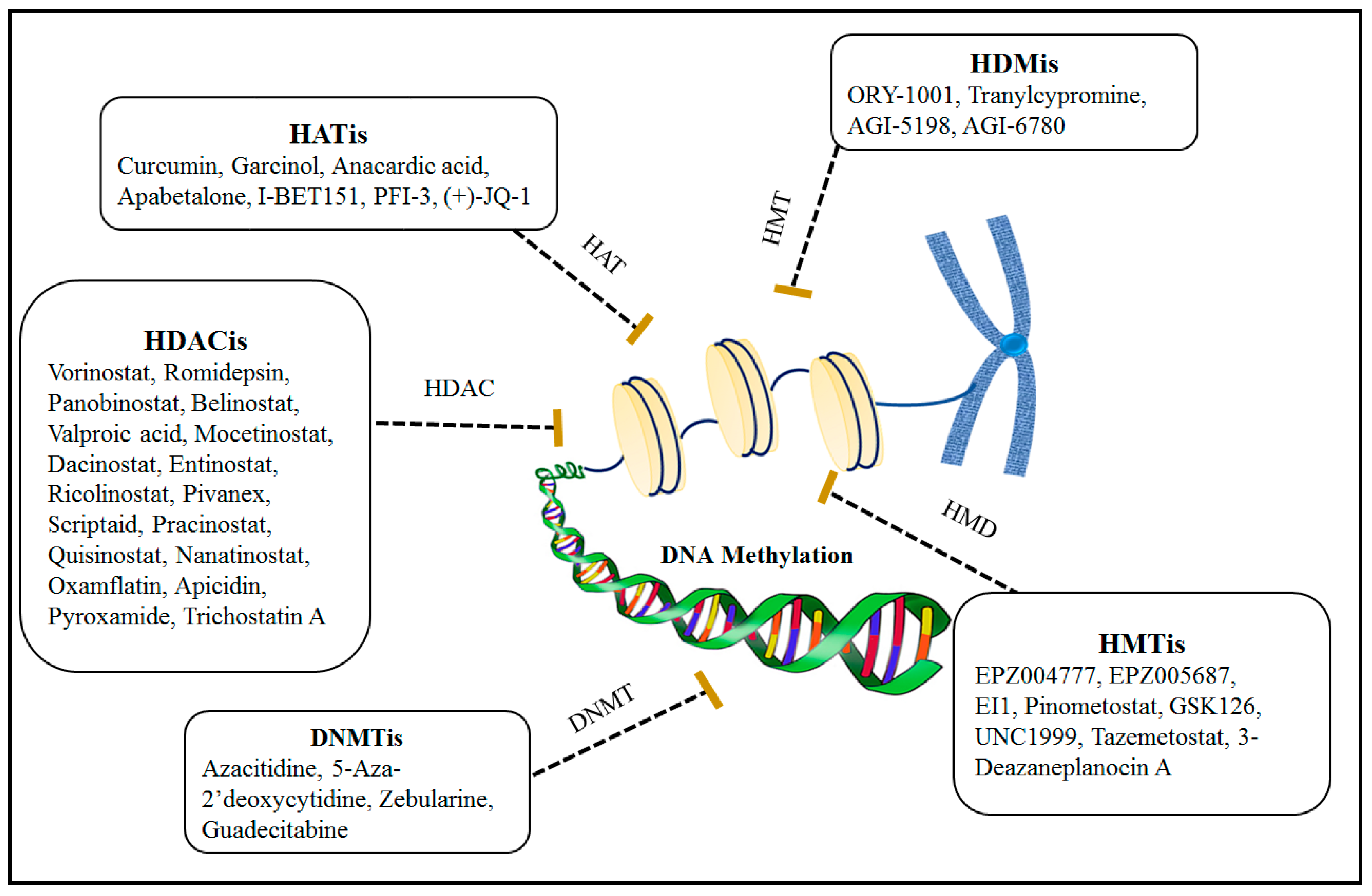
3. Epigenetic Therapy
3.1. Combination Effects of Using DNA Methylation and Histone Modification Inhibitors for Epigenetic Therapies
3.2. Potential Side Effects of Epigenetic Therapy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Study Highlights
References
- Noble, D. Conrad Waddington and the origin of epigenetics. J. Exp. Biol. 2015, 218 Pt 6, 816–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Recio, O.; Toro, M.A.; Bach, A. Past, present, and future of epigenetics applied to livestock breeding. Front. Genet. 2015, 6, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.-W.; Huang, K.; Yang, C.; Kang, C.-S. Non-coding RNAs as regulators in epigenetics. Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.L.; Grant, P.A. The role of DNA methylation and histone modifications in transcriptional regulation in humans. In Epigenetics: Development and Disease; Springer: Berlin/Heidelberg, Germany, 2013; pp. 289–317. [Google Scholar]
- Bohnsack, K.E.; Höbartner, C.; Bohnsack, M.T. Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: Mechanisms, cellular functions, and links to disease. Genes 2019, 10, 102. [Google Scholar] [CrossRef] [Green Version]
- Enroth, C.; Poulsen, L.D.; Iversen, S.; Kirpekar, F.; Albrechtsen, A.; Vinther, J. Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res. 2019, 47, e126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Wang, Q.; Huang, J.; Tang, C. Structural basis of N6-adenosine methylation by the METTL3–METTL14 complex. Nature 2016, 534, 575–578. [Google Scholar] [CrossRef]
- Wang, X.; Huang, J.; Zou, T.; Yin, P. Human m6A writers: Two subunits, 2 roles. RNA Biol. 2017, 14, 300–304. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Liu, Y.; Wu, R.; Bi, Z.; Yao, Y.; Liu, Q.; Wang, Y.; Wang, X. Understanding m6A function through uncovering the diversity roles of YTH domain-containing proteins. Mol. Biotechnol. 2019, 61, 355–364. [Google Scholar] [CrossRef]
- Schickel, R.; Boyerinas, B.; Park, S.; Peter, M. MicroRNAs: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008, 27, 5959–5974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Li, J.; Chen, R.; Gu, Q.; Yang, P.; Qian, W.; Ji, D.; Wang, Q.; Zhang, Z.; Tang, J. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.-S.; Liu, C.; Ma, H.; Dai, Q.; Sun, H.-L.; Luo, G.; Zhang, Z.; Zhang, L.; Hu, L.; Dong, X. Transcriptome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA. Mol. Cell 2019, 74, 1304–1316.e8. [Google Scholar] [CrossRef]
- Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Hendrick, A.; Andrews, B.; Webster, N.; Murat, P.; Mach, P.; Brandi, R.; Robson, S.C. METTL1 promotes let-7 MicroRNA processing via m7G methylation. Mol. Cell 2019, 74, 1278–1290.e9. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, A.M.; Chang, H.Y. Long noncoding RNAs in cancer pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Liao, S.E.; Ai, Y.; Fukunaga, R. RNA methyltransferase BCDIN3D is crucial for female fertility and miRNA and mRNA profiles in Drosophila ovaries. PLoS ONE 2019, 14, e0217603. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hou, D.; Chen, X.; Li, D.; Zhu, L.; Zhang, Y.; Li, J.; Bian, Z.; Liang, X.; Cai, X. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: Evidence of cross-kingdom regulation by microRNA. Cell Res. 2012, 22, 107–126. [Google Scholar] [CrossRef] [Green Version]
- Chenarani, N.; Emamjomeh, A.; Allahverdi, A.; Mirmostafa, S.; Afsharinia, M.H.; Zahiri, J. Bioinformatic tools for DNA methylation and histone modification: A survey. Genomics 2021, 113, 1098–1113. [Google Scholar] [CrossRef]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M. The human colon cancer methylome shows similar hypo-and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Allahverdi, A.; Yang, R.; Korolev, N.; Fan, Y.; Davey, C.A.; Liu, C.-F.; Nordenskiöld, L. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 2011, 39, 1680–1691. [Google Scholar] [CrossRef] [Green Version]
- Talbert, P.B.; Henikoff, S. Histone variants at a glance. J. Cell Sci. 2021, 134, jcs244749. [Google Scholar] [CrossRef]
- Ramazi, S.; Allahverdi, A.; Zahiri, J. Evaluation of post-translational modifications in histone proteins: A review on histone modification defects in developmental and neurological disorders. J. Biosci. 2020, 45, 135. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Weber, C.M.; Henikoff, S. Histone variants: Dynamic punctuation in transcription. Genes Dev. 2014, 28, 672–682. [Google Scholar] [CrossRef] [Green Version]
- Marques, M.; Laflamme, L.; Gervais, A.L.; Gaudreau, L. Reconciling the positive and negative roles of histone H2A.Z in gene transcription. Epigenetics 2010, 5, 267–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Zhi, D.; Aslibekyan, S.; Irvin, M.R.; Claas, S.A.; Borecki, I.B.; Ordovas, J.M.; Absher, D.M.; Arnett, D.K. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics 2013, 8, 802–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Li, Z.; Yu, D.; Wan, L.; Zhu, Y.; Lai, M.; Zhang, D. Polymorphisms involving gain or loss of CpG sites are significantly enriched in trait-associated SNPs. Oncotarget 2015, 6, 39995. [Google Scholar] [CrossRef] [Green Version]
- Heyn, H.; Moran, S.; Hernando-Herraez, I.; Sayols, S.; Gomez, A.; Sandoval, J.; Monk, D.; Hata, K.; Marques-Bonet, T.; Wang, L.; et al. DNA methylation contributes to natural human variation. Genome Res. 2013, 23, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Kilpinen, H.; Waszak, S.M.; Gschwind, A.R.; Raghav, S.K.; Witwicki, R.M.; Orioli, A.; Migliavacca, E.; Wiederkehr, M.; Gutierrez-Arcelus, M.; Panousis, N.I.; et al. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science 2013, 342, 744–747. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, J. MicroRNA-mediated breast cancer metastasis: From primary site to distant organs. Oncogene 2012, 31, 2499–2511. [Google Scholar] [CrossRef] [Green Version]
- Tanzer, M.; Balluff, B.; Distler, J.; Hale, K.; Leodolter, A.; Rocken, C.; Molnar, B.; Schmid, R.; Lofton-Day, C.; Schuster, T.; et al. Performance of epigenetic markers SEPT9 and ALX4 in plasma for detection of colorectal precancerous lesions. PLoS ONE 2010, 5, e9061. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lenferink, A.E.; Deng, Y.; Collins, C.; Cui, Q.; Purisima, E.O.; O’Connor-McCourt, M.D.; Wang, E. Corrigendum: Identification of high-quality cancer prognostic markers and metastasis network modules. Nat. Commun. 2012, 3, 655. [Google Scholar] [CrossRef] [Green Version]
- Kern, S.E. Why your new cancer biomarker may never work: Recurrent patterns and remarkable diversity in biomarker failures. Cancer Res. 2012, 72, 6097–6101. [Google Scholar] [CrossRef] [Green Version]
- Ginsburg, G.S.; Willard, H.F. Genomic and personalized medicine: Foundations and applications. Transl. Res. 2009, 154, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Zhou, H.; Fan, H.; Yuan, Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget 2017, 8, 110635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar] [CrossRef] [Green Version]
- Gujar, H.; Weisenberger, D.J.; Liang, G. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes 2019, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Kanai, Y.; Nakagawa, T.; Sakamoto, M.; Saito, H.; Ishii, H.; Hirohashi, S. Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int. J. Cancer. 2003, 105, 527–532. [Google Scholar] [CrossRef]
- Peng, D.F.; Kanai, Y.; Sawada, M.; Ushijima, S.; Hiraoka, N.; Kosuge, T.; Hirohashi, S. Increased DNA methyltransferase 1 (DNMT1) protein expression in precancerous conditions and ductal carcinomas of the pancreas. Cancer Sci. 2005, 96, 403–408. [Google Scholar] [CrossRef]
- Kanai, Y.; Ushijima, S.; Kondo, Y.; Nakanishi, Y.; Hirohashi, S. DNA methyltransferase expression and DNA methylation of CPG islands and peri-centromeric satellite regions in human colorectal and stomach cancers. Int. J. Cancer 2001, 91, 205–212. [Google Scholar] [CrossRef]
- Denis, H.; Ndlovu, M.N.; Fuks, F. Regulation of mammalian DNA methyltransferases: A route to new mechanisms. EMBO Rep. 2011, 12, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [Green Version]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113, 6411–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duursma, A.M.; Kedde, M.; Schrier, M.; le Sage, C.; Agami, R. miR-148 targets human DNMT3b protein coding region. RNA 2008, 14, 872–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braconi, C.; Huang, N.; Patel, T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology 2010, 51, 881–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López de Silanes, I.; Gorospe, M.; Taniguchi, H.; Abdelmohsen, K.; Srikantan, S.; Alaminos, M.; Berdasco, M.; Urdinguio, R.G.; Fraga, M.F.; Jacinto, F.V. The RNA-binding protein HuR regulates DNA methylation through stabilization of DNMT3b mRNA. Nucleic Acids Res. Spec. Publ. 2009, 37, 2658–2671. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Ozyerli-Goknar, E.; Bagci-Onder, T. Epigenetic deregulation of apoptosis in cancers. Cancers 2021, 13, 3210. [Google Scholar] [CrossRef]
- Young, N.L.; Dimaggio, P.A.; Garcia, B.A. The significance, development and progress of high-throughput combinatorial histone code analysis. Cell. Mol. Life Sci. 2010, 67, 3983–4000. [Google Scholar] [CrossRef]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W. A stem cell–like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, N.; Wong, J.; Tan, K.; Chung, H.; Yau, Y.; Cukuroglu, E.; Allahverdi, A.; Nordenskiöld, L.; Göke, J.; Geifman-Shochat, S. EZH2 promotes neoplastic transformation through VAV interaction-dependent extranuclear mechanisms. Oncogene 2018, 37, 461–477. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Cai, Y.; McGarvey, K.M.; Easwaran, H.; Van Neste, L.; Ohm, J.E.; O’Hagan, H.M.; Baylin, S.B. Polycomb CBX7 promotes initiation of heritable repression of genes frequently silenced with cancer-specific DNA hypermethylation. Cancer Res. 2009, 69, 6322–6330. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varier, R.A.; Timmers, H.T. Histone lysine methylation and demethylation pathways in cancer. Biochim. Biophys. Acta 2011, 1815, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Lorsbach, R.; Moore, J.; Mathew, S.; Raimondi, S.; Mukatira, S.; Downing, J. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t (10; 11)(q22; q23). Leukemia 2003, 17, 637–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poppe, B.; Vandesompele, J.; Schoch, C.; Lindvall, C.; Mrózek, K.; Bloomfield, C.D.; Beverloo, H.B.; Michaux, L.; Dastugue, N.; Herens, C. Expression analyses identify MLL as a prominent target of 11q23 amplification and support an etiologic role for MLL gain of function in myeloid malignancies. Blood 2004, 103, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Chen, A.; Yan, X.; Zhang, Y.; He, F.; Hayashi, Y.; Dong, Y.; Rao, Y.; Li, B.; Conway, R.M.; et al. Downregulation of RUNX1/CBFbeta by MLL fusion proteins enhances hematopoietic stem cell self-renewal. Blood 2014, 123, 1729–1738. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Archer, T.C.; Pomeroy, S.L. Medulloblastoma biology in the post-genomic era. Future Oncol. 2012, 8, 1597–1604. [Google Scholar] [CrossRef] [Green Version]
- Lederer, D.; Grisart, B.; Digilio, M.C.; Benoit, V.; Crespin, M.; Ghariani, S.C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am. J. Hum. Genet. 2012, 90, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.; Lan, F.; Bayliss, P.; de la Torre-Ubieta, L.; Huarte, M.; Qi, H.H.; Whetstine, J.R.; Bonni, A.; Roberts, T.M.; Shi, Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 2007, 128, 1077–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Ge, Z.; Wang, L.; Li, Q.; Wang, N.; Björkholm, M.; Jia, J.; Xu, D. The histone demethylase RBP2 Is overexpressed in gastric cancer and its inhibition triggers senescence of cancer cells. Gastroenterology 2010, 138, 981–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.C.; Lee, C.F.; Li, Y.S.; Chen, Y.R.; Hsiao, P.W.; Chan, M.Y.; Lin, F.M.; Huang, H.D.; Chen, Y.T.; Jeng, Y.M.; et al. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013, 73, 4711–4721. [Google Scholar] [CrossRef] [Green Version]
- Fattaey, A.R.; Helin, K.; Dembski, M.S.; Dyson, N.; Harlow, E.; Vuocolo, G.A.; Hanobik, M.G.; Haskell, K.M.; Oliff, A.; Defeo-Jones, D.; et al. Characterization of the retinoblastoma binding proteins RBP1 and RBP2. Oncogene 1993, 8, 3149–3156. [Google Scholar]
- Lu, P.J.; Sundquist, K.; Baeckstrom, D.; Poulsom, R.; Hanby, A.; Meier-Ewert, S.; Jones, T.; Mitchell, M.; Pitha-Rowe, P.; Freemont, P. A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J. Biol. Chem. 1999, 274, 15633–15645. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Zhu, Z.; Han, G.; Ye, X.; Xu, B.; Peng, Z.; Ma, Y.; Yu, Y.; Lin, H.; Chen, A.P. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 19226–19231. [Google Scholar] [CrossRef] [Green Version]
- Yamane, K.; Tateishi, K.; Klose, R.J.; Fang, J.; Fabrizio, L.A.; Erdjument-Bromage, H.; Taylor-Papadimitriou, J.; Tempst, P.; Zhang, Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell 2007, 25, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.G.; Nalls, M.A.; Gibbs, J.R.; Arepalli, S.; van der Brug, M.; Chong, S.; Moore, M.; Longo, D.L.; Cookson, M.R.; Traynor, B.J.; et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 2011, 20, 1164–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatz, M.; Pedersen, N.L.; Berg, S.; Johansson, B.; Johansson, K.; Mortimer, J.A.; Posner, S.F.; Viitanen, M.; Winblad, B.; Ahlbom, A. Heritability for Alzheimer’s disease: The study of dementia in Swedish twins. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52, M117–M125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Li, Y.; Camarillo, C.; Yao, Y.; Zhang, Y.; Xu, C.; Jiang, L. The anti-tumor histone deacetylase inhibitor SAHA and the natural flavonoid curcumin exhibit synergistic neuroprotection against amyloid-beta toxicity. PLoS ONE 2014, 9, e85570. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Bakulski, K.M.; Dolinoy, D.C.; Sartor, M.A.; Paulson, H.L.; Konen, J.R.; Lieberman, A.P.; Albin, R.L.; Hu, H.; Rozek, L.S. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J. Alzheimer’s Dis. 2012, 29, 571–588. [Google Scholar] [CrossRef] [Green Version]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef] [Green Version]
- Masliah, E.; Dumaop, W.; Galasko, D.; Desplats, P. Distinctive patterns of DNA methylation associated with Parkinson disease: Identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 2013, 8, 1030–1038. [Google Scholar] [CrossRef] [Green Version]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeid, R.; Schadt, A.; Dillmann, U.; Kostopoulos, P.; Fassbender, K.; Herrmann, W. Methylation status and neurodegenerative markers in Parkinson disease. Clin. Chem. 2009, 55, 1852–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldic, M.; Caruncho, H.; Liu, W.; Davis, J.; Satta, R.; Grayson, D.; Guidotti, A.; Costa, E. DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proc. Natl. Acad. Sci. USA 2004, 101, 348–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhubi, A.; Veldic, M.; Puri, N.; Kadriu, B.; Caruncho, H.; Loza, I.; Sershen, H.; Lajtha, A.; Smith, R.; Guidotti, A. An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophr. Res. 2009, 111, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.J.; Botuyan, M.V.; Wu, Y.; Ward, C.J.; Nicholson, G.A.; Hammans, S.; Hojo, K.; Yamanishi, H.; Karpf, A.R.; Wallace, D.C.; et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 2011, 43, 595–600. [Google Scholar] [CrossRef]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Urdinguio, R.G.; Sanchez-Mut, J.V.; Esteller, M. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol. 2009, 8, 1056–1072. [Google Scholar] [CrossRef]
- Amir, R.E.; Zoghbi, H.Y. Rett syndrome: Methyl-CpG-binding protein 2 mutations and phenotype-genotype correlations. Am. J. Med. Genet. 2000, 97, 147–152. [Google Scholar] [CrossRef]
- Ishii, T.; Makita, Y.; Ogawa, A.; Amamiya, S.; Yamamoto, M.; Miyamoto, A.; Oki, J. The role of different X-inactivation pattern on the variable clinical phenotype with Rett syndrome. Brain Dev. 2001, 23 (Suppl. 1), S161–S164. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Hou, J.; Maclean, A.; Nasir, J.; Lafuente, M.J.; Shu, X.; Kriaucionis, S.; Bird, A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. USA 2007, 104, 2709–2714. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, R.J.; Higgs, D.R. Molecular-clinical spectrum of the ATR-X syndrome. Am. J. Med. Genet. 2000, 97, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Kernohan, K.D.; Jiang, Y.; Tremblay, D.C.; Bonvissuto, A.C.; Eubanks, J.H.; Mann, M.R.; Berube, N.G. ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev. Cell 2010, 18, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Krantz, I.D. Cohesin and human disease. Annu. Rev. Genom. Hum. Genet. 2008, 9, 303–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulinsky, R.; Byrne, J.L.; Lowichik, A.; Viskochil, D.H. Fetus with interstitial del(5)(p13.1p14.2) diagnosed postnatally with Cornelia de Lange syndrome. Am. J. Med. Genet. Part A 2005, 137A, 336–338. [Google Scholar] [CrossRef]
- Lee, J.; Hagerty, S.; Cormier, K.A.; Kim, J.; Kung, A.L.; Ferrante, R.J.; Ryu, H. Monoallele deletion of CBP leads to pericentromeric heterochromatin condensation through ESET expression and histone H3 (K9) methylation. Hum. Mol. Genet. 2008, 17, 1774–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebel, V.I.; Kung, A.L.; Tanner, E.A.; Yang, H.; Bronson, R.T.; Livingston, D.M. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc. Natl. Acad. Sci. USA 2002, 99, 14789–14794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Wang, Y.; Liang, Y.; Zhao, M.; Long, H.; Ding, S.; Yin, H.; Lu, Q. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. 2011, 63, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Weaver, I.C.; Gauthier-Fisher, A.; Wang, H.; He, L.; Yeomans, J.; Wondisford, F.; Kaplan, D.R.; Miller, F.D. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev. Cell 2010, 18, 114–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugani, C.B.; Paquin, A.; Kaplan, D.R.; Miller, F.D. Coffin–Lowry syndrome: A role for RSK2 in mammalian neurogenesis. Dev. Biol. 2010, 347, 348–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Bogershausen, N.; Alanay, Y.; Simsek Kiper, P.O.; Plume, N.; Keupp, K.; Pohl, E.; Pawlik, B.; Rachwalski, M.; Milz, E.; et al. A mutation screen in patients with Kabuki syndrome. Hum. Genet. 2011, 130, 715–724. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Prathipati, P.; Pratiwi, R.; Phanus-Umporn, C.; Malik, A.A.; Schaduangrat, N.; Seenprachawong, K.; Wongchitrat, P.; Supokawej, A.; Prachayasittikul, V.; et al. Exploring the epigenetic drug discovery landscape. Expert Opin. Drug Discov. 2017, 12, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Wang, D.; Wang, D.; Jin, T.; Yang, L.; Wu, H.; Li, Y.; Zhao, J.; Du, F.; Song, M.; et al. HEDD: The human epigenetic drug database. Database 2016, 2016, baw159. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.; Zahnow, C.A.; Rudin, C.M.; Baylin, S.B. The future of epigenetic therapy in solid tumours—Lessons from the past. Nat. Rev. Clin. Oncol. 2013, 10, 256. [Google Scholar] [CrossRef]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, S.; Heil, O.; Lyko, F.; Brueckner, B. Azacytidine and decitabine induce gene-specific and non-random DNA demethylation in human cancer cell lines. PLoS ONE 2011, 6, e17388. [Google Scholar] [CrossRef]
- Goodyear, O.; Agathanggelou, A.; Novitzky-Basso, I.; Siddique, S.; McSkeane, T.; Ryan, G.; Vyas, P.; Cavenagh, J.; Stankovic, T.; Moss, P.; et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood 2010, 116, 1908–1918. [Google Scholar] [CrossRef] [Green Version]
- James, S.R.; Link, P.A.; Karpf, A.R. Epigenetic regulation of X-linked cancer/germline antigen genes by DNMT1 and DNMT3b. Oncogene 2006, 25, 6975–6985. [Google Scholar] [CrossRef] [Green Version]
- Kaminskas, E.; Farrell, A.; Abraham, S.; Baird, A.; Hsieh, L.S.; Lee, S.L.; Leighton, J.K.; Patel, H.; Rahman, A.; Sridhara, R.; et al. Approval summary: Azacitidine for treatment of myelodysplastic syndrome subtypes. Clin. Cancer Res. 2005, 11, 3604–3608. [Google Scholar] [CrossRef] [Green Version]
- Constantinides, P.G.; Jones, P.A.; Gevers, W. Functional striated muscle cells from non-myoblast precursors following 5-azacytidine treatment. Nature 1977, 267, 364–366. [Google Scholar] [CrossRef]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, A.; Halby, L.; Fahy, J.; Arimondo, P.B. Targeting DNA methylation with small molecules: What’s next? Miniperspective. J. Med. Chem. 2015, 58, 2569–2583. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.; Huang, T.H.; Brown, R.; Nephew, K.P. The epigenetics of ovarian cancer drug resistance and resensitization. Am. J. Obstet. Gynecol. 2004, 191, 1552–1572. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Zahnow, C.A.; Ahuja, N.; Baylin, S.B. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016, 76, 1683–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Li, L.; Liu, X.; Wang, D.; Tu, B.; Wang, L.; Wang, H.; Zhu, W.G. 5-Aza-2′-deoxycytidine reactivates gene expression via degradation of pRb pocket proteins. FASEB J. 2012, 26, 449–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balch, C.; Yan, P.; Craft, T.; Young, S.; Skalnik, D.G.; Huang, T.H.; Nephew, K.P. Antimitogenic and chemosensitizing effects of the methylation inhibitor zebularine in ovarian cancer. Mol. Cancer Ther. 2005, 4, 1505–1514. [Google Scholar] [CrossRef] [Green Version]
- Chuang, J.C.; Warner, S.L.; Vollmer, D.; Vankayalapati, H.; Redkar, S.; Bearss, D.J.; Qiu, X.; Yoo, C.B.; Jones, P.A. S110, a 5-Aza-2′-deoxycytidine–containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol. Cancer Ther. 2010, 9, 1443–1450. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.J.; Roboz, G.; Rizzieri, D.; Jabbour, E.; Stock, W.; O’Connell, C.; Yee, K.; Tibes, R.; Griffiths, E.A.; Walsh, K.; et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015, 16, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, P.; Paluch, B.E.; Matsuzaki, J.; James, S.R.; Collamat-Lai, G.; Karbach, J.; Nemeth, M.J.; Taverna, P.; Karpf, A.R.; Griffiths, E.A. Immunomodulatory action of SGI-110, a hypomethylating agent, in acute myeloid leukemia cells and xenografts. Leuk. Res. 2014, 38, 1332–1341. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Li, H.-Q.; Liu, F. DNA methyltransferase inhibitors and their therapeutic potential. Curr. Top. Med. Chem. 2018, 18, 2448–2457. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Griffiths, E.A.; Roboz, G.J.; Busque, L.; Wells, R.A.; Odenike, O.; Steensma, D.P.; Yee, K.W.; Faderl, S.; Amrein, P.C. A phase 2 dose-confirmation study of oral ASTX727, a combination of oral decitabine with a cytidine deaminase inhibitor (CDAi) cedazuridine (E7727), in subjects with myelodysplastic syndromes (MDS). Blood 2017, 130, 4274. [Google Scholar]
- Segura-Pacheco, B.; Trejo-Becerril, C.; Perez-Cardenas, E.; Taja-Chayeb, L.; Mariscal, I.; Chavez, A.; Acuña, C.; Salazar, A.M.; Lizano, M.; Dueñas-Gonzalez, A. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin. Cancer Res. 2003, 9, 1596–1603. [Google Scholar]
- Lee, W.J.; Shim, J.Y.; Zhu, B.T. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol. 2005, 68, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Sun, X.; Xie, Y.; Zhuang, Y.; Yao, R.; Xu, K. Anti-proliferative activity of HPOB against multiple myeloma cells via p21 transcriptional activation. Molecules 2018, 23, 1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominique, G.H.; Arimondo, P.B. Challenges in developing novel DNA methyltransferase inhibitors for cancer therapy. Future Med. Chem. 2014, 6, 1237–1240. [Google Scholar] [CrossRef]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Waters, N.J. Preclinical pharmacokinetics and pharmacodynamics of pinometostat (EPZ-5676), a first-in-class, small molecule S-adenosyl methionine competitive inhibitor of DOT1L. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 891–901. [Google Scholar] [CrossRef]
- Siu, L.L.; Rasco, D.W.; Vinay, S.P.; Romano, P.M.; Menis, J.; Opdam, F.L.; Heinhuis, K.M.; Egger, J.L.; Gorman, S.; Parasrampuria, R. METEOR-1: A phase I study of GSK3326595, a first-in-class protein arginine methyltransferase 5 (PRMT5) inhibitor, in advanced solid tumours. Ann. Oncol. 2019, 30, v159. [Google Scholar] [CrossRef]
- Tremblay-LeMay, R.; Rastgoo, N.; Pourabdollah, M.; Chang, H. EZH2 as a therapeutic target for multiple myeloma and other haematological malignancies. Biomark. Res. 2018, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Tang, A.; Castoreno, A.; Kuo, S.; Wang, Q.; Kuballa, P.; Xavier, R.; Shamji, A.; Schreiber, S.; Wagner, B. Gossypol and an HMT G9a inhibitor act in synergy to induce cell death in pancreatic cancer cells. Cell Death Dis. 2013, 4, e690. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, M.; Hori, M.; Fujikawa, D.; Ohsugi, T.; Honma, D.; Adachi, N.; Katano, H.; Hishima, T.; Kobayashi, S.; Nakano, K. Targeting excessive EZH1 and EZH2 activities for abnormal histone methylation and transcription network in malignant lymphomas. Cell. Rep. 2019, 29, 2321–2337.e7. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Ciesielski, O.; Balcerczyk, A. The methylation status of the epigenome: Its emerging role in the regulation of tumor angiogenesis and tumor growth, and potential for drug targeting. Cancers 2018, 10, 268. [Google Scholar] [CrossRef] [Green Version]
- Weiss, M.C.; Agulnik, M. Tazemetostat as a treatment for epithelioid sarcoma. Expert Opin. Orphan Drugs 2020, 8, 311–315. [Google Scholar] [CrossRef]
- Cimmino, L.; Abdel-Wahab, O.; Levine, R.L.; Aifantis, I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell 2011, 9, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, N.; George, T.L.; Otterson, G.A.; Verschraegen, C.; Wen, H.; Carbone, D.; Herman, J.; Bertino, E.M.; He, K. Advances in epigenetic therapeutics with focus on solid tumors. Clin. Epigenetics 2021, 13, 83. [Google Scholar] [CrossRef]
- Cole, P.A. Chemical probes for histone-modifying enzymes. Nat. Chem. Biol. 2008, 4, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, A.; Banerjee, S.; Stafford, L.J.; Xia, C.; Liu, M.; Aggarwal, B.B. Curcumin-induced suppression of cell proliferation correlates with down-regulation of cyclin D1 expression and CDK4-mediated retinoblastoma protein phosphorylation. Oncogene 2002, 21, 8852–8861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, A.-J.; Jiang, G.; Li, L.-T.; Zheng, J.-N. Curcumin induces apoptosis through mitochondrial pathway and caspases activation in human melanoma cells. Mol. Biol. Rep. 2015, 42, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galdeano, C.; Ciulli, A. Selectivity on-target of bromodomain chemical probes by structure-guided medicinal chemistry and chemical biology. Future Med. Chem. 2016, 8, 1655–1680. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Puri, R.; Wolski, K.; Ballantyne, C.M.; Barter, P.J.; Brewer, H.B.; Kastelein, J.J.; Hu, B.; Uno, K.; Kataoka, Y.; et al. Effect of the BET Protein Inhibitor, RVX-208, on Progression of Coronary Atherosclerosis: Results of the Phase 2b, Randomized, Double-Blind, Multicenter, ASSURE Trial. Am. J. Cardiovasc. Drugs 2016, 16, 55–65. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Zhu, W.-G. Molecular mechanisms of epigenetic regulators as activatable targets in cancer theranostics. Curr. Med. Chem. 2019, 26, 1328–1350. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, W.G. Targeting histone deacetylases for cancer therapy: From molecular mechanisms to clinical implications. Int. J. Biol. Sci. 2014, 10, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Mrakovcic, M.; Fröhlich, L.F. Molecular determinants of cancer therapy resistance to HDAC inhibitor-induced autophagy. Cancers 2019, 12, 109. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, S. Drugs targeting epigenetic modifications and plausible therapeutic strategies against colorectal cancer. Front. Pharmacol. 2019, 10, 588. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non–small-cell lung cancer. J. Clin. Oncol. 2010, 28, 56. [Google Scholar] [CrossRef] [Green Version]
- Poole, R.M. Belinostat: First global approval. Drugs 2014, 74, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 2014, 5, 5637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410. [Google Scholar] [CrossRef] [Green Version]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [Green Version]
- García-Gutiérrez, L.; Delgado, M.D.; León, J. MYC oncogene contributions to release of cell cycle brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef] [Green Version]
- Pathania, R.; Ramachandran, S.; Mariappan, G.; Thakur, P.; Shi, H.; Choi, J.H.; Manicassamy, S.; Kolhe, R.; Prasad, P.D.; Sharma, S.; et al. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-like Cells and Attenuates Mammary Tumor Growth. Cancer Res. 2016, 76, 3224–3235. [Google Scholar] [CrossRef] [Green Version]
- Kala, R.; Peek, G.W.; Hardy, T.M.; Tollefsbol, T.O. MicroRNAs: An emerging science in cancer epigenetics. J. Clin. Bioinform. 2013, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Saito, H.; Liang, G.; Friedman, J.M. Epigenetic alterations and microRNA misexpression in cancer and autoimmune diseases: A critical review. Clin. Rev. Allergy Immunol. 2014, 47, 128–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huaying, C.; Xing, J.; Luya, J.; Linhui, N.; Di, S.; Xianjun, D. A signature of five long non-coding rnas for predicting the prognosis of Alzheimer’s disease based on competing endogenous RNA networks. Front. Aging Neurosci. 2021, 12, 598606. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.H.; Yang, F.; Chen, B.F.; Lu, Z.; Huo, X.S.; Zhou, W.P.; Wang, F.; Sun, S.H. The histone deacetylase 4/SP1/microrna-200a regulatory network contributes to aberrant histone acetylation in hepatocellular carcinoma. Hepatology 2011, 54, 2025–2035. [Google Scholar] [CrossRef] [PubMed]
- Schubert, J.; Brabletz, T. p53 Spreads out further: Suppression of EMT and stemness by activating miR-200c expression. Cell Res. 2011, 21, 705–707. [Google Scholar] [CrossRef]
- Rhodes, L.V.; Tate, C.R.; Segar, H.C.; Burks, H.E.; Phamduy, T.B.; Hoang, V.; Elliott, S.; Gilliam, D.; Pounder, F.N.; Anbalagan, M.; et al. Suppression of triple-negative breast cancer metastasis by pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT master regulators. Breast Cancer Res. Treat. 2014, 145, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Wendtner, C.M. Cocktail of eternity: HDAC meets miR. Blood 2012, 119, 1095–1096. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Sun, M.; Zhou, S.; Guo, B. Class I HDAC inhibitor mocetinostat induces apoptosis by activation of miR-31 expression and suppression of E2F6. Cell Death Discov. 2016, 2, 16036. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, M.; Calin, G.A. Epigenetics and miRNAs in human cancer. Adv. Genet. 2010, 70, 87–99. [Google Scholar]
- Brocks, D.; Schmidt, C.R.; Daskalakis, M.; Jang, H.S.; Shah, N.M.; Li, D.; Li, J.; Zhang, B.; Hou, Y.; Laudato, S. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet. 2017, 49, 1052–1060. [Google Scholar] [CrossRef]
- Mekala, J.R.; Naushad, S.M.; Ponnusamy, L.; Arivazhagan, G.; Sakthiprasad, V.; Pal-Bhadra, M. Epigenetic regulation of miR-200 as the potential strategy for the therapy against triple-negative breast cancer. Gene 2018, 641, 248–258. [Google Scholar] [CrossRef]
- Xiao, W.; Zhou, Q.; Wen, X.; Wang, R.; Liu, R.; Wang, T.; Shi, J.; Hu, Y.; Hou, J. Small-Molecule Inhibitors Overcome Epigenetic Reprogramming for Cancer Therapy. Front. Pharmacol. 2021, 12, 702360. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Toh, H.C.; Chow, P.; Chung, A.Y.; Meyers, D.J.; Cole, P.A.; Ooi, L.L.; Lee, C.G. MicroRNA-224 is up-regulated in hepatocellular carcinoma through epigenetic mechanisms. FASEB J. 2012, 26, 3032–3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Ansarullah; Kumar, D.; Jaggi, M.; Chauhan, S.C. Targeting microRNAs in pancreatic cancer: Microplayers in the big game. Cancer Res. 2013, 73, 6541–6547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Cao, M.; Zhang, J.; Hu, K.; Yin, Z.; Zhou, Z.; Xiao, X.; Yang, Y.; Sheng, W.; Wu, Y.; et al. Hyaluronic acid-chitosan nanoparticles for co-delivery of MiR-34a and doxorubicin in therapy against triple negative breast cancer. Biomaterials 2014, 35, 4333–4344. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, L.; Alvarez-Errico, D.; Esteller, M. The Contribution of Epigenetics to Cancer Immunotherapy. Trends Immunol. 2020, 41, 676–691. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Q.; Chen, C.S.; Chen, J.J.; Zhou, L.P.; Xu, H.L.; Jin, W.W.; Wu, J.B.; Gao, S.M. Histone deacetylases inhibitor trichostatin A increases the expression of Dleu2/miR-15a/16-1 via HDAC3 in non-small cell lung cancer. Mol. Cell. Biochem. 2013, 383, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, C. HDAC2-mediated proliferation of trophoblast cells requires the miR-183/FOXA1/IL-8 signaling pathway. J. Cell. Physiol. 2021, 236, 2544–2558. [Google Scholar] [CrossRef]
- Qi, W.; Chan, H.; Teng, L.; Li, L.; Chuai, S.; Zhang, R.; Zeng, J.; Li, M.; Fan, H.; Lin, Y.; et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 21360–21365. [Google Scholar] [CrossRef] [Green Version]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; Macnevin, C.J.; Liu, F.; Gao, C.; Huang, X.P.; Kuznetsova, E.; et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef]
- Chen, Z.; Du, Y.; Liu, X.; Chen, H.; Weng, X.; Guo, J.; Wang, M.; Wang, X.; Wang, L. EZH2 inhibition suppresses bladder cancer cell growth and metastasis via the JAK2/STAT3 signaling pathway. Oncol. Lett. 2019, 18, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Guo, Y.; Zhang, X.; Li, J.; Li, L.; Zhang, S.; Shan, C. ORY-1001 suppresses cell growth and induces apoptosis in lung cancer through triggering HK2 mediated Warburg effect. Front. Pharmacol. 2018, 9, 1411. [Google Scholar] [CrossRef]
- Sun, Q.; Ding, D.; Liu, X.; Guo, S.W. Tranylcypromine, a lysine-specific demethylase 1 (LSD1) inhibitor, suppresses lesion growth and improves generalized hyperalgesia in mouse with induced endometriosis. Reprod. Biol. Endocrinol. 2016, 14, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieling, H.; Bleich, S. Tranylcypromine: New perspectives on an “old” drug. Eur. Arch. Psychiatry Clin. Neurosci. 2006, 256, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Botman, D.; Smits, M.A.; Hira, V.V.; van Lith, S.A.; Stap, J.; Henneman, P.; Khurshed, M.; Lenting, K.; Mul, A.N.; et al. Radioprotection of IDH1-Mutated Cancer Cells by the IDH1-Mutant Inhibitor AGI-5198. Cancer Res. 2015, 75, 4790–4802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Chaidos, A.; Caputo, V.; Gouvedenou, K.; Liu, B.; Marigo, I.; Chaudhry, M.S.; Rotolo, A.; Tough, D.F.; Smithers, N.N.; Bassil, A.K.; et al. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood 2014, 123, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Fan, D.; Wang, Y. The BET bromodomain inhibitor i-BET151 impairs ovarian cancer metastasis and improves antitumor immunity. Cell Tissue Res. 2018, 374, 577–585. [Google Scholar] [CrossRef]
- Vangamudi, B.; Paul, T.A.; Shah, P.K.; Kost-Alimova, M.; Nottebaum, L.; Shi, X.; Zhan, Y.; Leo, E.; Mahadeshwar, H.S.; Protopopov, A. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: Insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res. 2015, 75, 3865–3878. [Google Scholar] [CrossRef] [Green Version]
- Briere, D.; Sudhakar, N.; Woods, D.M.; Hallin, J.; Engstrom, L.D.; Aranda, R.; Chiang, H.; Sodre, A.L.; Olson, P.; Weber, J.S.; et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol. Immunother. 2018, 67, 381–392. [Google Scholar] [CrossRef]
- Atadja, P.; Gao, L.; Kwon, P.; Trogani, N.; Walker, H.; Hsu, M.; Yeleswarapu, L.; Chandramouli, N.; Perez, L.; Versace, R.; et al. Selective growth inhibition of tumor cells by a novel histone deacetylase inhibitor, NVP-LAQ824. Cancer Res. 2004, 64, 689–695. [Google Scholar] [CrossRef] [Green Version]
- Romanski, A.; Schwarz, K.; Keller, M.; Wietbrauk, S.; Vogel, A.; Roos, J.; Oancea, C.; Brill, B.; Kramer, O.H.; Serve, H.; et al. Deacetylase inhibitors modulate proliferation and self-renewal properties of leukemic stem and progenitor cells. Cell Cycle 2012, 11, 3219–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.; Valone, F.; Lipera, W.; Irwin, D.; Paroly, W.; Natale, R.; Sreedharan, S.; Keer, H.; Lum, B.; Scappaticci, F.; et al. Phase II trial of the histone deacetylase inhibitor pivaloyloxymethyl butyrate (Pivanex, AN-9) in advanced non-small cell lung cancer. Lung Cancer 2004, 45, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, M.J.; Naushad, S.M.; Lavanya, A.; Srinivas, C.; Devi, T.A.; Sampathkumar, S.; Dharan, D.B.; Bhadra, M.P. Scriptaid cause histone deacetylase inhibition and cell cycle arrest in HeLa cancer cells: A study on structural and functional aspects. Gene 2017, 627, 379–386. [Google Scholar] [CrossRef]
- Meng, Q.; Yang, G.; Yang, Y.; Ding, F.; Hu, F. Protective effects of histone deacetylase inhibition by Scriptaid on brain injury in neonatal rat models of cerebral ischemia and hypoxia. Int. J. Clin. Exp. Pathol. 2020, 13, 179–191. [Google Scholar]
- Abaza, Y.M.; Kadia, T.M.; Jabbour, E.J.; Konopleva, M.Y.; Borthakur, G.; Ferrajoli, A.; Estrov, Z.; Wierda, W.G.; Alfonso, A.; Chong, T.H.; et al. Phase 1 dose escalation multicenter trial of pracinostat alone and in combination with azacitidine in patients with advanced hematologic malignancies. Cancer 2017, 123, 4851–4859. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.L.; Peng, C.Y.; Lai, M.J.; Chen, C.H.; Lee, H.Y.; Wang, J.C.; Liou, J.P.; Pan, S.L.; Teng, C.M. Novel oral histone deacetylase inhibitor, MPT0E028, displays potent growth-inhibitory activity against human B-cell lymphoma in vitro and in vivo. Oncotarget 2015, 6, 4976–4991. [Google Scholar] [CrossRef] [Green Version]
- Arts, J.; King, P.; Mariën, A.; Floren, W.; Beliën, A.; Janssen, L.; Pilatte, I.; Roux, B.; Decrane, L.; Gilissen, R. JNJ-26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin. Cancer Res. 2009, 15, 6841–6851. [Google Scholar] [CrossRef] [Green Version]
- Moffat, D.; Patel, S.; Day, F.; Belfield, A.; Donald, A.; Rowlands, M.; Wibawa, J.; Brotherton, D.; Stimson, L.; Clark, V.; et al. Discovery of 2-(6-{[(6-fluoroquinolin-2-yl)methyl]amino}bicyclo[3.1.0]hex-3-yl)-N-hydroxypyrim idine-5-carboxamide (CHR-3996), a class I selective orally active histone deacetylase inhibitor. J. Med. Chem. 2010, 53, 8663–8678. [Google Scholar] [CrossRef]
- Faghihloo, E.; Araei, Y.; Mohammadi, M.; Mirzaei, H.; Mohammadi, H.R.; Mokhtari-Azad, T. The effect of oxamflatin on the E-cadherin expression in gastric cancer cell line. Cancer Gene Ther. 2016, 23, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Ahn, S.H.; Kim, Y.K.; Bae, G.U.; Yoon, J.W.; Hong, S.; Lee, H.Y.; Lee, Y.W.; Lee, H.W.; Han, J.W. Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J. Biol. Chem. 2002, 277, 2073–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, L.M.; Webb, Y.; Agus, D.B.; Higgins, B.; Tolentino, T.R.; Kutko, M.C.; LaQuaglia, M.P.; Drobnjak, M.; Cordon-Cardo, C.; Scher, H.I.; et al. Inhibition of transformed cell growth and induction of cellular differentiation by pyroxamide, an inhibitor of histone deacetylase. Clin. Cancer Res. 2001, 7, 962–970. [Google Scholar] [PubMed]
- Vigushin, D.M.; Ali, S.; Pace, P.E.; Mirsaidi, N.; Ito, K.; Adcock, I.; Coombes, R.C. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 2001, 7, 971–976. [Google Scholar]
- Jahangeer, S.; Elliott, R.M.; Henneberry, R.C. beta-Adrenergic receptor induction in HeLa cells: Synergistic effect of 5-azacytidine and butyrate. Biochem. Biophys. Res. Commun. 1982, 108, 1434–1440. [Google Scholar] [CrossRef]
- Ginder, G.D.; Whitters, M.J.; Pohlman, J.K. Activation of a chicken embryonic globin gene in adult erythroid cells by 5-azacytidine and sodium butyrate. Proc. Natl. Acad. Sci. USA 1984, 81, 3954–3958. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Gabrielson, E.; Chen, W.; Anbazhagan, R.; van Engeland, M.; Weijenberg, M.P.; Herman, J.G.; Baylin, S.B. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet. 2002, 31, 141–149. [Google Scholar] [CrossRef]
- Yamashita, K.; Upadhyay, S.; Osada, M.; Hoque, M.O.; Xiao, Y.; Mori, M.; Sato, F.; Meltzer, S.J.; Sidransky, D. Pharmacologic unmasking of epigenetically silenced tumor suppressor genes in esophageal squamous cell carcinoma. Cancer Cell 2002, 2, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Belinsky, S.A.; Klinge, D.M.; Stidley, C.A.; Issa, J.P.; Herman, J.G.; March, T.H.; Baylin, S.B. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res. 2003, 63, 7089–7093. [Google Scholar]
- Claus, R.; Lubbert, M. Epigenetic targets in hematopoietic malignancies. Oncogene 2003, 22, 6489–6496. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef]
- Plumb, J.A.; Strathdee, G.; Sludden, J.; Kaye, S.B.; Brown, R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000, 60, 6039–6044. [Google Scholar] [PubMed]
- Karpf, A.R.; Jones, D.A. Reactivating the expression of methylation silenced genes in human cancer. Oncogene 2002, 21, 5496–5503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.; Salgaller, M.; Samid, D.; Johnson, B.; Herlyn, M.; Lassam, N.; Treisman, J.; Rosenberg, S. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2′-deoxycytidine. Cancer Res. 1994, 54, 1766–1771. [Google Scholar]
- Liang, G.; Gonzales, F.A.; Jones, P.A.; Orntoft, T.F.; Thykjaer, T. Analysis of gene induction in human fibroblasts and bladder cancer cells exposed to the methylation inhibitor 5-aza-2′-deoxycytidine. Cancer Res. 2002, 62, 961–966. [Google Scholar] [PubMed]
- Mohandas, T.; Sparkes, R.S.; Shapiro, L.J. Reactivation of an inactive human X chromosome: Evidence for X inactivation by DNA methylation. Science 1981, 211, 393–396. [Google Scholar] [CrossRef]
- Eversole-Cire, P.; Ferguson-Smith, A.C.; Sasaki, H.; Brown, K.D.; Cattanach, B.M.; Gonzales, F.A.; Surani, M.A.; Jones, P.A. Activation of an imprinted Igf 2 gene in mouse somatic cell cultures. Mol. Cell. Biol. 1993, 13, 4928–4938. [Google Scholar] [CrossRef]
- Jackson-Grusby, L.; Laird, P.W.; Magge, S.N.; Moeller, B.J.; Jaenisch, R. Mutagenicity of 5-aza-2′-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc. Natl. Acad. Sci. USA 1997, 94, 4681–4685. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.S.; Estecio, M.R.; Garcia-Manero, G.; Kantarjian, H.M.; Issa, J.-P.J. Comment on “Chromosomal instability and tumors promoted by DNA hypomethylation” and “Induction of tumors in mice by genomic hypomethylation”. Science 2003, 302, 1153. [Google Scholar] [CrossRef] [Green Version]
- Karpf, A.R.; Moore, B.C.; Ririe, T.O.; Jones, D.A. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2′-deoxycytidine. Mol. Pharmacol. 2001, 59, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Peterson, E.J.; Bogler, O.; Taylor, S.M. p53-mediated repression of DNA methyltransferase 1 expression by specific DNA binding. Cancer Res. 2003, 63, 6579–6582. [Google Scholar] [PubMed]
- Jüttermann, R.; Li, E.; Jaenisch, R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. USA 1994, 91, 11797–11801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathrop, M.; Gut, I.; Heath, S.; Tost, J.; Gress, T.; Hudson, T. International network of cancer genome projects (The International Cancer Genome Consortium). Nat. Dig. 2010, 464, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutvagner, G.; Zamore, P.D. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002, 297, 2056–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; De Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic enhancement of cancer therapy using HDAC inhibitors: Opportunity for clinical trials. Front. Genet. 2020, 11, 578011. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Warych, A.; Szoszkiewicz, M. Novel approaches to epigenetic therapies: From drug combinations to epigenetic editing. Genes 2021, 12, 208. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kundukad, B.; Allahverdi, A.; Nordensköld, L.; Doyle, P.S.; van der Maarel, J.R. Gelation of the genome by topoisomerase II targeting anticancer agents. Soft Matter 2013, 9, 1656–1663. [Google Scholar] [CrossRef]
- Knox, T.; Sahakian, E.; Banik, D.; Hadley, M.; Palmer, E.; Noonepalle, S.; Kim, J.; Powers, J.; Gracia-Hernandez, M.; Oliveira, V. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci. Rep. 2019, 9, 6136. [Google Scholar] [CrossRef] [Green Version]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Guerrero, E.; Götz, R.; Doose, S.; Sauer, M.; Rodríguez-Gil, A.; Nerreter, T.; Kortüm, K.M.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M. Upregulation of CD38 expression on multiple myeloma cells by novel HDAC6 inhibitors is a class effect and augments the efficacy of daratumumab. Leukemia 2021, 35, 201–214. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories | Epi-Drug Name | Alternate Name | Conditions | Formula | FDA Approved | Ref. |
|---|---|---|---|---|---|---|
| DNMTi | Azacitidine | 5-azacitidine | AML, CML and MDS | Yes | [120,137] | |
| 5-Aza-2′-deoxycytidine | Decitabine | AML, CML and MDS | Yes | [123] | ||
| Zebularine | NSC309132 | Cancers | No | [127] | ||
| Decitabine | ASTX727 | AML, CML and MDS | No | [133] | ||
| Epigallocatechin gallate | EGCG | Cancers | No | [136] | ||
| Guadecitabine | SGI-110 | Cancers | No | [128,129] |
| Categories | Epi-Drug Name | Alternate Name | Conditions | Formula | FDA Approved | Ref. |
|---|---|---|---|---|---|---|
| HMTi | EPZ004777 | ----------- | MLL-translocated leukemia | No | [138] | |
| EPZ015938 | Pemrametostat | Cancers, Hematologic malignancies | No | [141] | ||
| BIX-01294 | ----------- | Cancers | No | [146] | ||
| UNC0638 | ----------- | Cancers, anti-viral | No | [146] | ||
| BRD4770 | ----------- | Cancers | No | [144] | ||
| EPZ005687 | ----------- | MM | C32H37N5O3 | No | [142] | |
| EI1 | KB-145943 | Large and follicular B-cell lymphomas, Cancers | No | [189] | ||
| Pinometostat | EPZ5676 | AML, ALL, MDS | No | [139] | ||
| GSK126 | GSK2816126A | MM | No | [142] | ||
| UNC1999 | ----------- | Cancers, Hematologic malignancies | C33H43N7O2 | No | [190,191] | |
| Tazemetostat | EPZ-6438 | Advance epithelioid sarcoma | Yes | [147] | ||
| 3-Deazaneplanocin A | DZNEP | Cancers | No | [143] | ||
| HDMi | ORY-1001 | ----------- | Cancers and AML | No | [192] | |
| Tranylcypromine * | Dl-Tranylcypromine | Depression and endometriosis | C9H12NO2S0.5 | No * | [193,194] | |
| Enasidenib | AG-221 | AML | Yes | [149] | ||
| AGI-5198 | IDH-C35 | Cancers | No | [195] | ||
| AGI-6780 | ----------- | AML and cancers | No | [196] | ||
| HATi | Curcumin | Diferuloylmethane | Cancers, MM | No | [152] | |
| Garcinol | Garcinia gummi-gutta fruit | Cancers | No | [112] | ||
| Anacardic acid | Hydroginkgolic acid | Cancers | No | [112] | ||
| Apabetalone | RVX-208 | Diabetes Atherosclerosis | No | [156] | ||
| I-BET151 | GSK1210151A | Cancers, MM | No | [197,198] | ||
| PFI-3 | ----------- | Cancers and lymphoma | C19H19N3O2 | No | [199] | |
| (+)-JQ-1 | JQ1 | Cancers | No | [154] | ||
| HDACi | Vorinostat | SAHA | CTCL | Yes | [115] | |
| Romidepsin | Depsipeptide | CTCL and PTCL | Yes | [166,167] | ||
| Panobinostat | LBH589 | MM | Yes | [163] | ||
| Belinostat | PXD-101 | PTCL | Yes | [162] | ||
| Valproic acid ** | Sodium valproate | Seizures, cancers | No | [168] | ||
| Mocetinostat | MGCD0103 | Cancers, MDS | No | [200] | ||
| Dacinostat | LAQ824 | Cancers and AML | No | [201,202] | ||
| Entinostat | MS-275 | Cancers, Hematologic malignancies | No | [203] | ||
| Ricolinostat | ACY-1215 | Cancers and MM | No | [204] | ||
| Pivanex | AN-9 | Cancers | No | [205] | ||
| Scriptaid | GCK 1026 | Cancers and TBI | No | [206,207] | ||
| Pracinostat | SB939 | Cancers, Hematologic malignancies | No | [208] | ||
| givinostat | ITF-2357 | Cancers | No | [165] | ||
| Resminostat | RAS2410 | CTCL | No | [165] | ||
| abexinostat | PCI-24781 | Cancers, Hematologic malignancies | No | [165] | ||
| MPT0E028 | ----------- | B-cell lymphomas, Cancers | No | [209] | ||
| Quisinostat | JNJ-26481585 | Cancers, Hematologic malignancies | No | [210] | ||
| Nanatinostat | CHR-3996 | Cancers | No | [211] | ||
| Oxamflatin | Metacept-3 | Cancers | No | [212] | ||
| Apicidin | OSI 2040 | Cancers | No | [213] | ||
| Pyroxamide | ----------- | Cancers, Hematologic malignancies | No | [214] | ||
| Trichostatin A | TSA | Cancers | No | [215] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahafnejad, Z.; Ramazi, S.; Allahverdi, A. An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes 2023, 14, 873. https://doi.org/10.3390/genes14040873
Sahafnejad Z, Ramazi S, Allahverdi A. An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes. 2023; 14(4):873. https://doi.org/10.3390/genes14040873
Chicago/Turabian StyleSahafnejad, Zahra, Shahin Ramazi, and Abdollah Allahverdi. 2023. "An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review" Genes 14, no. 4: 873. https://doi.org/10.3390/genes14040873
APA StyleSahafnejad, Z., Ramazi, S., & Allahverdi, A. (2023). An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes, 14(4), 873. https://doi.org/10.3390/genes14040873