
Specific Deoxyceramide Species Correlate with Expression of Macular Telangiectasia Type 2 (MacTel2) in a SPTLC2 Carrier HSAN1 Family


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
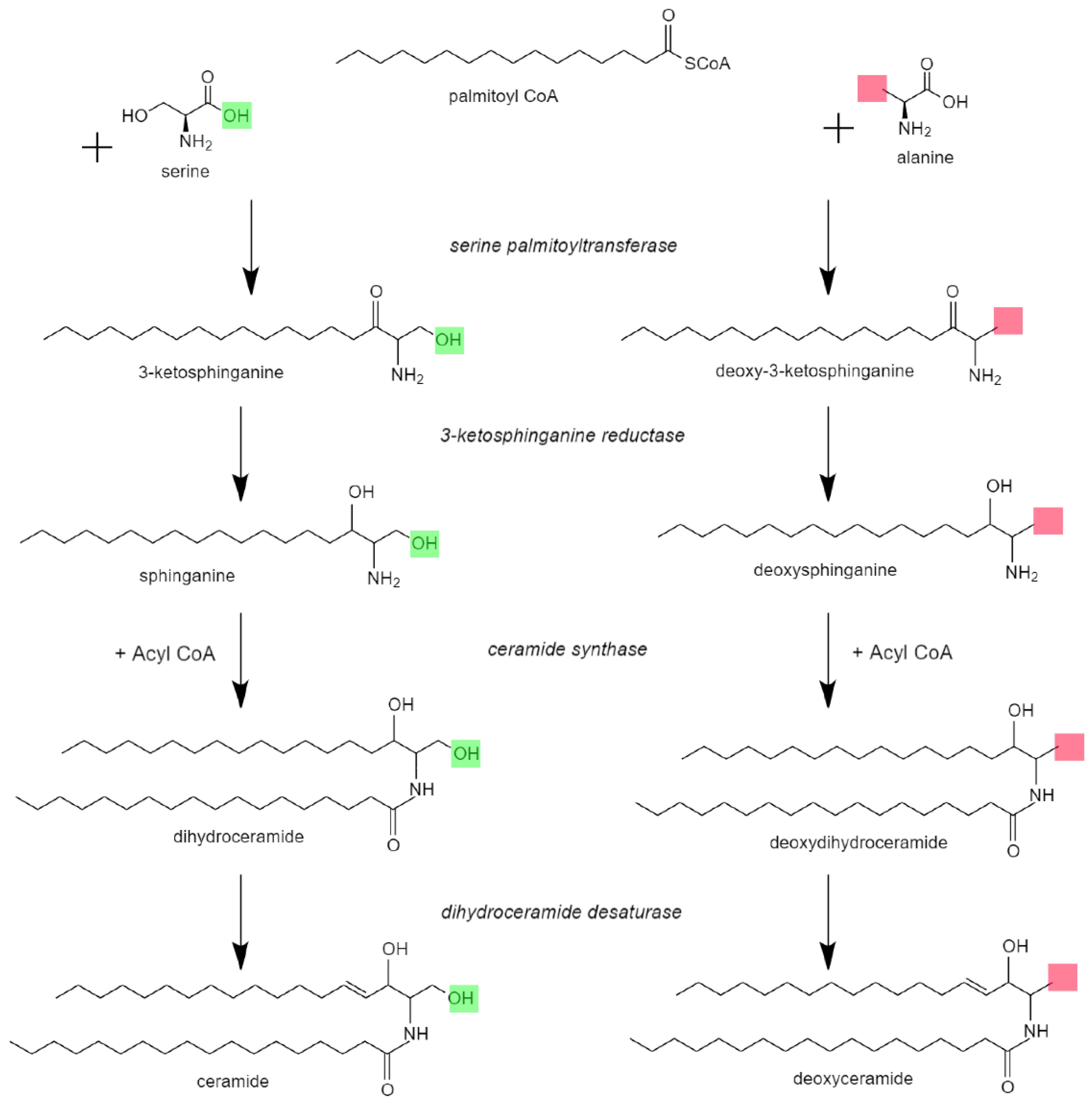
:1. Introduction
2. Materials and Methods
2.1. DNA Sequencing and Patient Consent
2.2. Sample Collection and Lipidomic Analysis
2.3. Literature Review
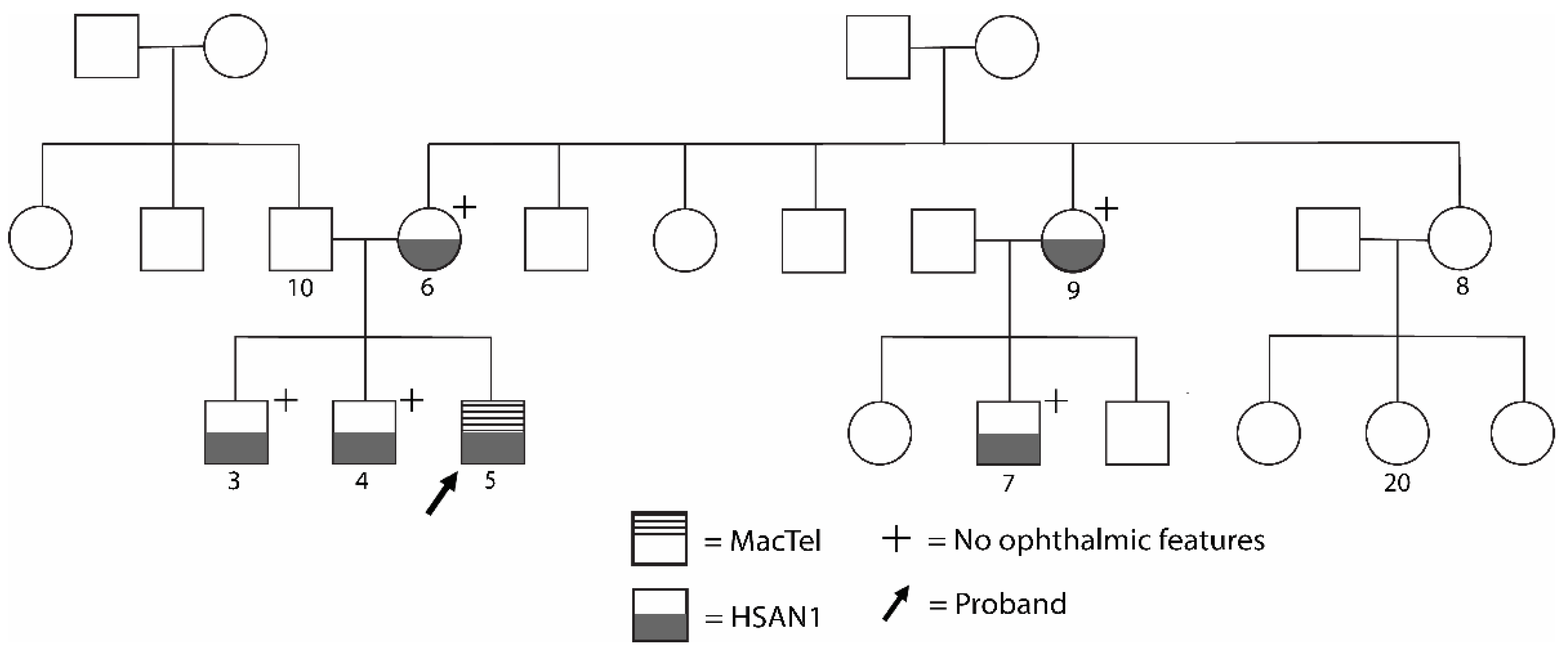
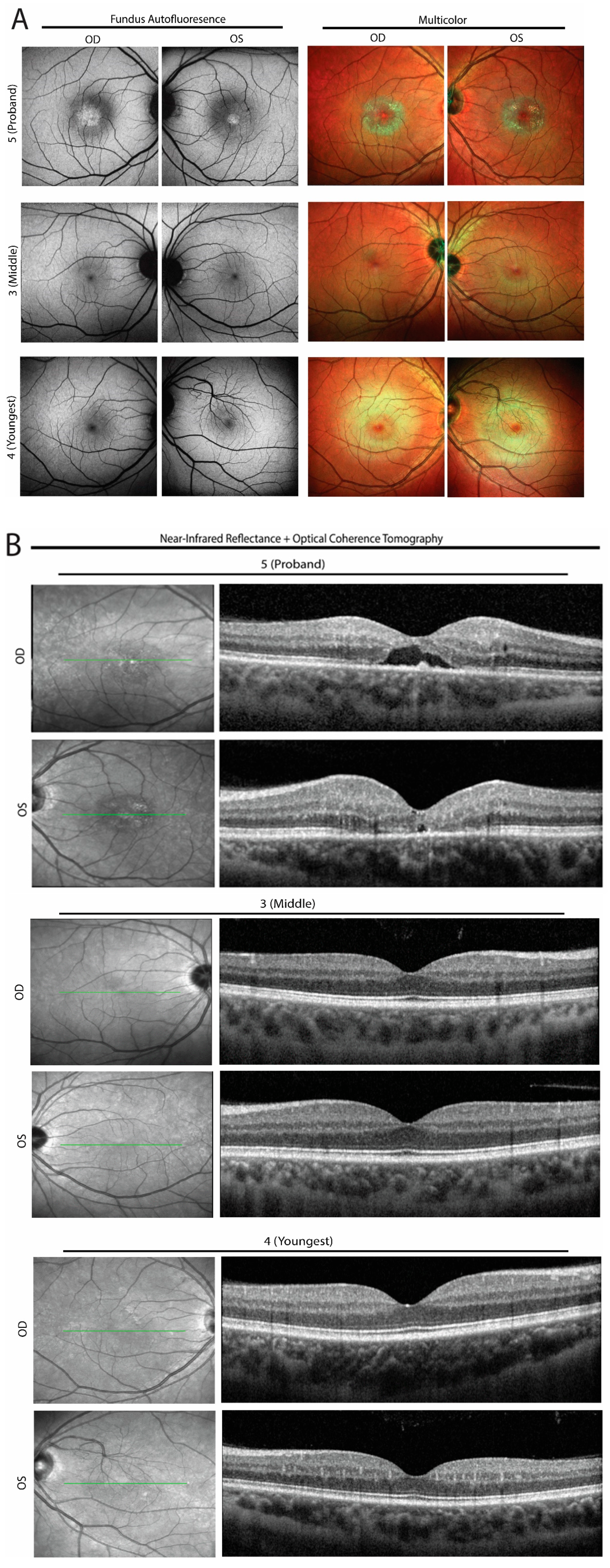
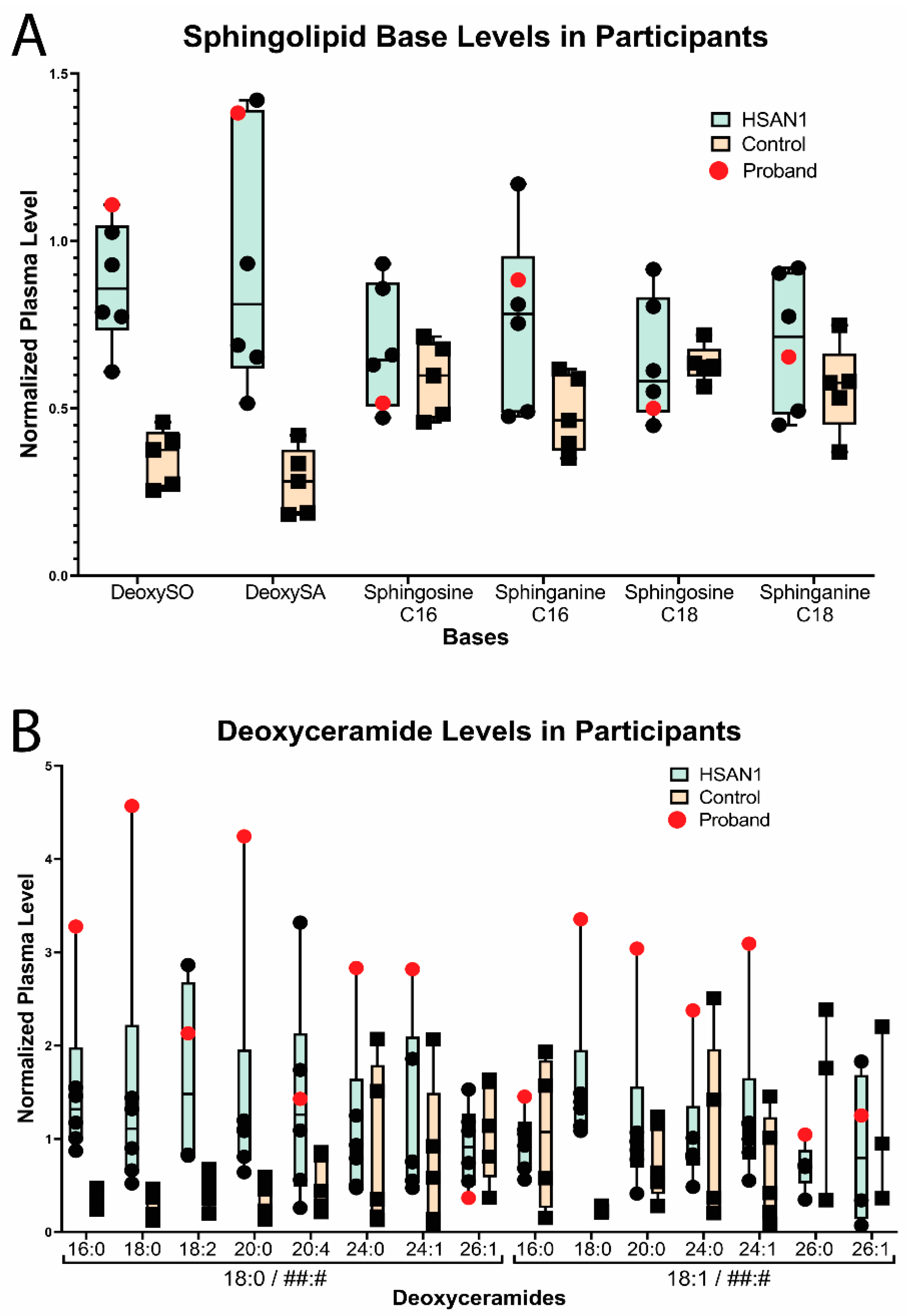
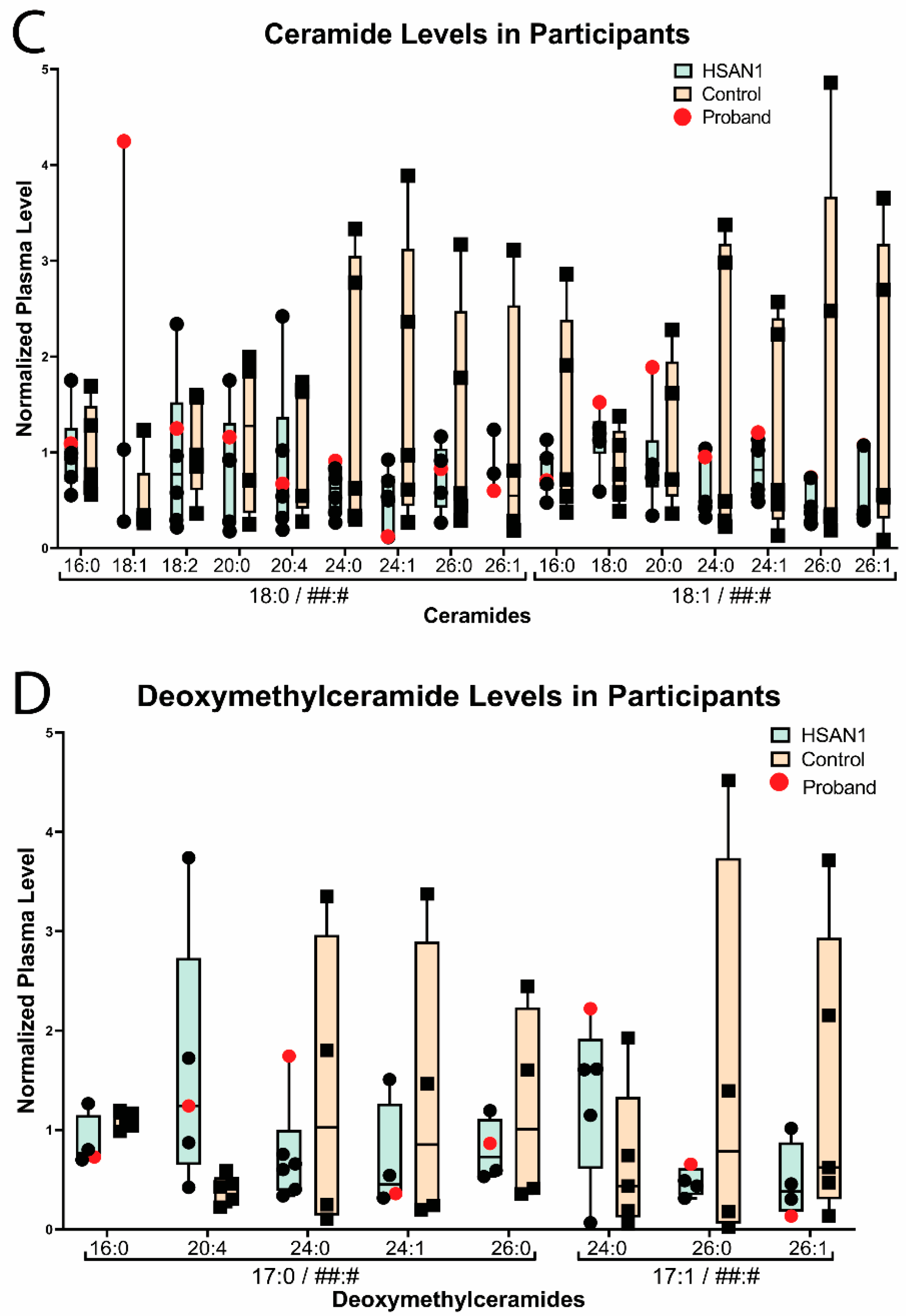
3. Familial Case Report
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Auer-Grumbach, M. Chapter 50—Hereditary Sensory and Autonomic Neuropathies. In Handbook of Clinical Neurology; Said, G., Krarup, C., Eds.; Peripheral Nerve Disorders; Elsevier: Amsterdam, The Netherlands, 2013; Volume 115, pp. 893–906. [Google Scholar]
- Wang, Y.; Niu, Y.; Zhang, Z.; Gable, K.; Gupta, S.D.; Somashekarappa, N.; Han, G.; Zhao, H.; Myasnikov, A.G.; Kalathur, R.C.; et al. Structural Insights into the Regulation of Human Serine Palmitoyltransferase Complexes. Nat. Struct. Mol. Biol. 2021, 28, 240–248. [Google Scholar] [CrossRef]
- Hornemann, T.; Wei, Y.; von Eckardstein, A. Is the Mammalian Serine Palmitoyltransferase a High-Molecular-Mass Complex? Biochem. J. 2007, 405, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Ikushiro, H.; Islam, M.M.; Okamoto, A.; Hoseki, J.; Murakawa, T.; Fujii, S.; Miyahara, I.; Hayashi, H. Structural Insights into the Enzymatic Mechanism of Serine Palmitoyltransferase from Sphingobacterium Multivorum. J. Biochem. 2009, 146, 549–562. [Google Scholar] [CrossRef]
- Harrison, P.J.; Dunn, T.M.; Campopiano, D.J. Sphingolipid Biosynthesis in Man and Microbes. Nat. Prod. Rep. 2018, 35, 921–954. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, G.A. SPTLC1-Related Hereditary Sensory Neuropathy. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Penno, A.; Reilly, M.M.; Houlden, H.; Laurá, M.; Rentsch, K.; Niederkofler, V.; Stoeckli, E.T.; Nicholson, G.; Eichler, F.; Brown, R.H.; et al. Hereditary Sensory Neuropathy Type 1 Is Caused by the Accumulation of Two Neurotoxic Sphingolipids. J. Biol. Chem. 2010, 285, 11178–11187. [Google Scholar] [CrossRef]
- Rotthier, A.; Auer-Grumbach, M.; Janssens, K.; Baets, J.; Penno, A.; Almeida-Souza, L.; Van Hoof, K.; Jacobs, A.; De Vriendt, E.; Schlotter-Weigel, B.; et al. Mutations in the SPTLC2 Subunit of Serine Palmitoyltransferase Cause Hereditary Sensory and Autonomic Neuropathy Type I. Am. J. Hum. Genet. 2010, 87, 513–522. [Google Scholar] [CrossRef]
- Bode, H.; Bourquin, F.; Suriyanarayanan, S.; Wei, Y.; Alecu, I.; Othman, A.; Von Eckardstein, A.; Hornemann, T. HSAN1 Mutations in Serine Palmitoyltransferase Reveal a Close Structure-Function-Phenotype Relationship. Hum. Mol. Genet. 2016, 25, 853–865. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [CrossRef]
- Gantner, M.L.; Eade, K.; Wallace, M.; Handzlik, M.K.; Fallon, R.; Trombley, J.; Bonelli, R.; Giles, S.; Harkins-Perry, S.; Heeren, T.F.C.; et al. Serine and Lipid Metabolism in Macular Disease and Peripheral Neuropathy. N. Engl. J. Med. 2019, 381, 1422–1433. [Google Scholar] [CrossRef]
- Gomes Rodrigues, F.; Pipis, M.; Heeren, T.F.C.; Fruttiger, M.; Gantner, M.; Vermeirsch, S.; Okada, M.; Friedlander, M.; Reilly, M.M.; Egan, C. Description of a Patient Cohort with Hereditary Sensory Neuropathy Type 1 without Retinal Disease Macular Telangiectasia Type 2—Implications for Retinal Screening in HSN1. J. Peripher. Nerv. Syst. 2022, 27, 215–224. [Google Scholar] [CrossRef]
- Triplett, J.; Yiannikas, C.; Nicholson, G.; Sue, C.; Hornemann, T. Hereditary Sensory and Autonomic Neuropathy Type IC Accompanied by Upper Motor Neuron Abnormalities and Type II Juxtafoveal Retinal Telangiectasias. J. Peripher. Nerv. Syst. 2019, 24, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Kedarisetti, K.C.; Narayanan, R.; Stewart, M.W.; Reddy Gurram, N.; Khanani, A.M. Macular Telangiectasia Type 2: A Comprehensive Review. Clin. Ophthalmol. Auckl. NZ 2022, 16, 3297–3309. [Google Scholar] [CrossRef] [PubMed]
- Sinha, T.; Ikelle, L.; Naash, M.I.; Al-Ubaidi, M.R. The Intersection of Serine Metabolism and Cellular Dysfunction in Retinal Degeneration. Cells 2020, 9, 674. [Google Scholar] [CrossRef] [PubMed]
- Eade, K.; Gantner, M.L.; Hostyk, J.A.; Nagasaki, T.; Giles, S.; Fallon, R.; Harkins-Perry, S.; Baldini, M.; Lim, E.W.; Scheppke, L.; et al. Serine Biosynthesis Defect Due to Haploinsufficiency of PHGDH Causes Retinal Disease. Nat. Metab. 2021, 3, 366–377. [Google Scholar] [CrossRef]
- Bonelli, R.; Jackson, V.E.; Prasad, A.; Munro, J.E.; Farashi, S.; Heeren, T.F.C.; Pontikos, N.; Scheppke, L.; Friedlander, M.; Egan, C.A.; et al. Identification of Genetic Factors Influencing Metabolic Dysregulation and Retinal Support for MacTel, a Retinal Disorder. Commun. Biol. 2021, 4, 274. [Google Scholar] [CrossRef]
- Bonelli, R.; Woods, S.M.; Ansell, B.R.E.; Heeren, T.F.C.; Egan, C.A.; Khan, K.N.; Guymer, R.; Trombley, J.; Friedlander, M.; Bahlo, M.; et al. Systemic Lipid Dysregulation Is a Risk Factor for Macular Neurodegenerative Disease. Sci. Rep. 2020, 10, 12165. [Google Scholar] [CrossRef]
- Bonelli, R.; Ansell, B.R.E.; Lotta, L.; Scerri, T.; Clemons, T.E.; Leung, I.; Peto, T.; Bird, A.C.; Sallo, F.B.; Langenberg, C.; et al. Genetic Disruption of Serine Biosynthesis Is a Key Driver of Macular Telangiectasia Type 2 Aetiology and Progression. Genome Med. 2021, 13, 39. [Google Scholar] [CrossRef]
- Saba, S.; Chen, Y.; Maddipati, K.R.; Hackett, M.; Hu, B.; Li, J. Demyelination in Hereditary Sensory Neuropathy Type-1C. Ann. Clin. Transl. Neurol. 2020, 7, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Gaver, R.C.; Sweeley, C.C. Methods for Methanolysis of Sphingolipids and Direct Determination of Long-Chain Bases by Gas Chromatography. J. Am. Oil Chem. Soc. 1965, 42, 294–298. [Google Scholar] [CrossRef]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An Illustrator for the Presentation and Visualization of Biological Sequences: Figure 1. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef]
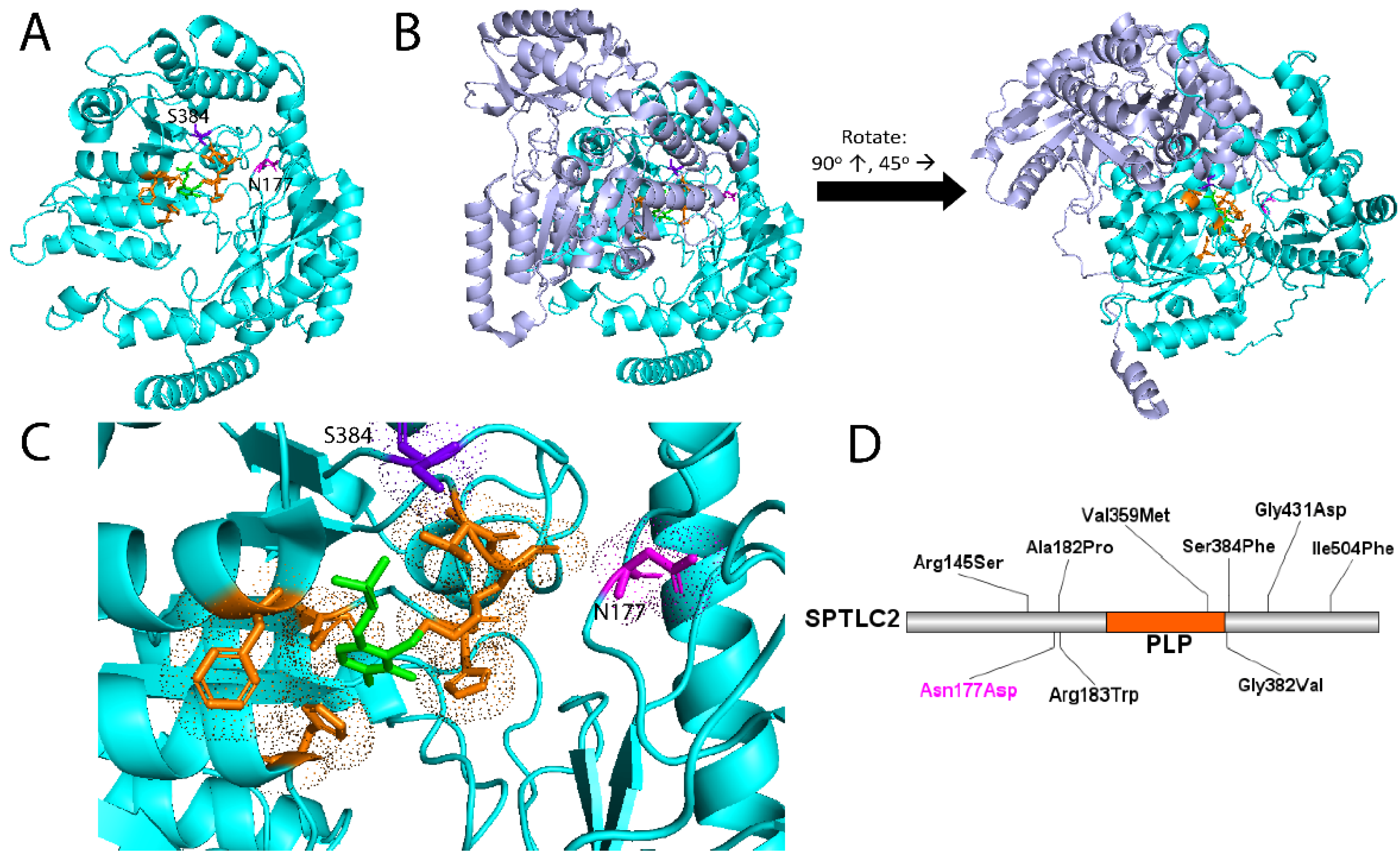
- Suriyanarayanan, S.; Othman, A.; Dräger, B.; Schirmacher, A.; Young, P.; Mulahasanovic, L.; Hörtnagel, K.; Biskup, S.; von Eckardstein, A.; Hornemann, T.; et al. A Novel Variant (Asn177Asp) in SPTLC2 Causing Hereditary Sensory Autonomic Neuropathy Type 1C. NeuroMolecular Med. 2019, 21, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Heeren, T.F.C.; Holz, F.G.; Charbel Issa, P. First Symptoms and Their Age of Onset in Macular Telangiectasia Type 2. Retina 2014, 34, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, H.-A.; Yoon, Y.H. OCT Angiography Findings of Tamoxifen Retinopathy: Similarity with Macular Telangiectasia Type 2. Ophthalmol. Retina 2019, 3, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.; Park, Y.J.; Kim, H.-A.; Holz, F.G.; Issa, P.C.; Yoon, Y.H.; Tzaridis, S. Tamoxifen Retinopathy and Macular Telangiectasia Type 2: Similarities and Differences on Multimodal Retinal Imaging. Ophthalmol. Retin. 2022, 7, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, S.; Petri, B.J.; Kruer, T.; Green, B.; Dougherty, S.; Wittliff, J.L.; Klinge, C.M.; Clem, B.F. Serine Synthesis Influences Tamoxifen Response in ER+ Human Breast Carcinoma. Endocr. Relat. Cancer 2021, 28, 27–37. [Google Scholar] [CrossRef]
- Esaki, K.; Sayano, T.; Sonoda, C.; Akagi, T.; Suzuki, T.; Ogawa, T.; Okamoto, M.; Yoshikawa, T.; Hirabayashi, Y.; Furuya, S. L-Serine Deficiency Elicits Intracellular Accumulation of Cytotoxic Deoxysphingolipids and Lipid Body Formation. J. Biol. Chem. 2015, 290, 14595–14609. [Google Scholar] [CrossRef] [PubMed]
- Hammad, S.M.; Baker, N.L.; El Abiad, J.M.; Spassieva, S.D.; Pierce, J.S.; Rembiesa, B.; Bielawski, J.; Lopes-Virella, M.F.; Klein, R.L.; DCCT/EDIC Group of Investigators. Increased Plasma Levels of Select Deoxy-Ceramide and Ceramide Species Are Associated with Increased Odds of Diabetic Neuropathy in Type 1 Diabetes: A Pilot Study. Neuromolecular Med. 2017, 19, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Handzlik, M.K.; Gengatharan, J.M.; Frizzi, K.E.; McGregor, G.H.; Martino, C.; Rahman, G.; Gonzalez, A.; Moreno, A.M.; Green, C.R.; Guernsey, L.S.; et al. Insulin-Regulated Serine and Lipid Metabolism Drive Peripheral Neuropathy. Nature 2023, 614, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Semler, A.; Hammad, S.; Lopes-Virella, M.F.; Klein, R.L.; Huang, Y. Deoxysphingolipids Upregulate MMP-1, Downregulate TIMP-1, and Induce Cytotoxicity in Human Schwann Cells. Neuromolecular Med. 2022, 24, 352–362. [Google Scholar] [CrossRef]
- Bryan, J.M.; Fufa, T.D.; Bharti, K.; Brooks, B.P.; Hufnagel, R.B.; McGaughey, D.M. Identifying Core Biological Processes Distinguishing Human Eye Tissues with Precise Systems-Level Gene Expression Analyses and Weighted Correlation Networks. Hum. Mol. Genet. 2018, 27, 3325–3339. [Google Scholar] [CrossRef]
- Voigt, A.P.; Whitmore, S.S.; Lessing, N.D.; DeLuca, A.P.; Tucker, B.A.; Stone, E.M.; Mullins, R.F.; Scheetz, T.E. Spectacle: An Interactive Resource for Ocular Single-Cell RNA Sequencing Data Analysis. Exp. Eye Res. 2020, 200, 108204. [Google Scholar] [CrossRef] [PubMed]
- Othman, A.; Bianchi, R.; Alecu, I.; Wei, Y.; Porretta-Serapiglia, C.; Lombardi, R.; Chiorazzi, A.; Meregalli, C.; Oggioni, N.; Cavaletti, G.; et al. Lowering Plasma 1-Deoxysphingolipids Improves Neuropathy in Diabetic Rats. Diabetes 2015, 64, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Kugathasan, U.; Baskozos, G.; Priestman, D.A.; Fugger, N.; Lone, M.A.; Othman, A.; Chu, K.H.; Blesneac, I.; Wilson, E.R.; et al. An IPSC Model of Hereditary Sensory Neuropathy-1 Reveals L-Serine-Responsive Deficits in Neuronal Ganglioside Composition and Axoglial Interactions. Cell Rep. Med. 2021, 2, 100345. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A.; Chaurasia, B.; Holland, W.L. Metabolic Messengers: Ceramides. Nat. Metab. 2019, 1, 1051–1058. [Google Scholar] [CrossRef]
- Ueda, N. Ceramide-Induced Apoptosis in Renal Tubular Cells: A Role of Mitochondria and Sphingosine-1-Phoshate. Int. J. Mol. Sci. 2015, 16, 5076–5124. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Hayakawa, T.; Makino, A.; Iwamoto, K.; Ito, K.; Sato, S.B.; Kobayashi, T. Long Chain Ceramides Raise the Main Phase Transition of Monounsaturated Phospholipids to Physiological Temperature. Sci. Rep. 2022, 12, 20803. [Google Scholar] [CrossRef]
- Montes, L.R.; Ruiz-Argüello, M.B.; Goñi, F.M.; Alonso, A. Membrane Restructuring via Ceramide Results in Enhanced Solute Efflux. J. Biol. Chem. 2002, 277, 11788–11794. [Google Scholar] [CrossRef]
- Chen, H.; Tran, J.-T.A.; Brush, R.S.; Saadi, A.; Rahman, A.K.; Yu, M.; Yasumura, D.; Matthes, M.T.; Ahern, K.; Yang, H.; et al. Ceramide Signaling in Retinal Degeneration. Adv. Exp. Med. Biol. 2012, 723, 553–558. [Google Scholar] [CrossRef]
- Duan, J.; Merrill, A.H. 1-Deoxysphingolipids Encountered Exogenously and Made de Novo: Dangerous Mysteries inside an Enigma. J. Biol. Chem. 2015, 290, 15380–15389. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Saavedra, V.; Mangan, M.S.J.; Penno, A.; Thiele, C.; Latz, E.; Kuerschner, L. 1-Deoxysphingolipids Cause Autophagosome and Lysosome Accumulation and Trigger NLRP3 Inflammasome Activation. Autophagy 2021, 17, 1947–1961. [Google Scholar] [CrossRef] [PubMed]
- Toft-Kehler, A.K.; Skytt, D.M.; Svare, A.; Lefevere, E.; Van Hove, I.; Moons, L.; Waagepetersen, H.S.; Kolko, M. Mitochondrial Function in Müller Cells—Does It Matter? Mitochondrion 2017, 36, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Kady, N.M.; Liu, X.; Lydic, T.A.; Syed, M.H.; Navitskaya, S.; Wang, Q.; Hammer, S.S.; O’Reilly, S.; Huang, C.; Seregin, S.S.; et al. ELOVL4-Mediated Production of Very Long-Chain Ceramides Stabilizes Tight Junctions and Prevents Diabetes-Induced Retinal Vascular Permeability. Diabetes 2018, 67, 769–781. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Brown, S.H.J.; Lim, X.Y.; Fiveash, C.E.; Osborne, B.; Bentley, N.L.; Braude, J.P.; Mitchell, T.W.; Coster, A.C.F.; Don, A.S.; et al. Regulation of Glucose Homeostasis and Insulin Action by Ceramide Acyl-Chain Length: A Beneficial Role for Very Long-Chain Sphingolipid Species. Biochim. Biophys. Acta 2016, 1861, 1828–1839. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-H.; Jang, W.-H.; Seo, J.A.; Park, M.; Lee, T.R.; Park, Y.-H.; Kim, D.K.; Lim, K.-M. Decrease of Ceramides with Very Long-Chain Fatty Acids and Downregulation of Elongases in a Murine Atopic Dermatitis Model. J. Investig. Dermatol. 2012, 132, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, A.; Westerberg, R.; Jacobsson, A. Fatty Acid Elongases in Mammals: Their Regulation and Roles in Metabolism. Prog. Lipid Res. 2006, 45, 237–249. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, L.M.Q.; Saba, S.; Li, J.; Prasov, L.; Miller, J.M.L. Specific Deoxyceramide Species Correlate with Expression of Macular Telangiectasia Type 2 (MacTel2) in a SPTLC2 Carrier HSAN1 Family. Genes 2023, 14, 931. https://doi.org/10.3390/genes14040931
Wilson LMQ, Saba S, Li J, Prasov L, Miller JML. Specific Deoxyceramide Species Correlate with Expression of Macular Telangiectasia Type 2 (MacTel2) in a SPTLC2 Carrier HSAN1 Family. Genes. 2023; 14(4):931. https://doi.org/10.3390/genes14040931
Chicago/Turabian StyleWilson, Lindsey M. Q., Sadaf Saba, Jun Li, Lev Prasov, and Jason M. L. Miller. 2023. "Specific Deoxyceramide Species Correlate with Expression of Macular Telangiectasia Type 2 (MacTel2) in a SPTLC2 Carrier HSAN1 Family" Genes 14, no. 4: 931. https://doi.org/10.3390/genes14040931
APA StyleWilson, L. M. Q., Saba, S., Li, J., Prasov, L., & Miller, J. M. L. (2023). Specific Deoxyceramide Species Correlate with Expression of Macular Telangiectasia Type 2 (MacTel2) in a SPTLC2 Carrier HSAN1 Family. Genes, 14(4), 931. https://doi.org/10.3390/genes14040931