
Identification and Molecular Characterization of a Novel Large-Scale Variant (Exons 4_18 Loss) in the LDLR Gene as a Cause of Familial Hypercholesterolaemia in an Italian Family

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
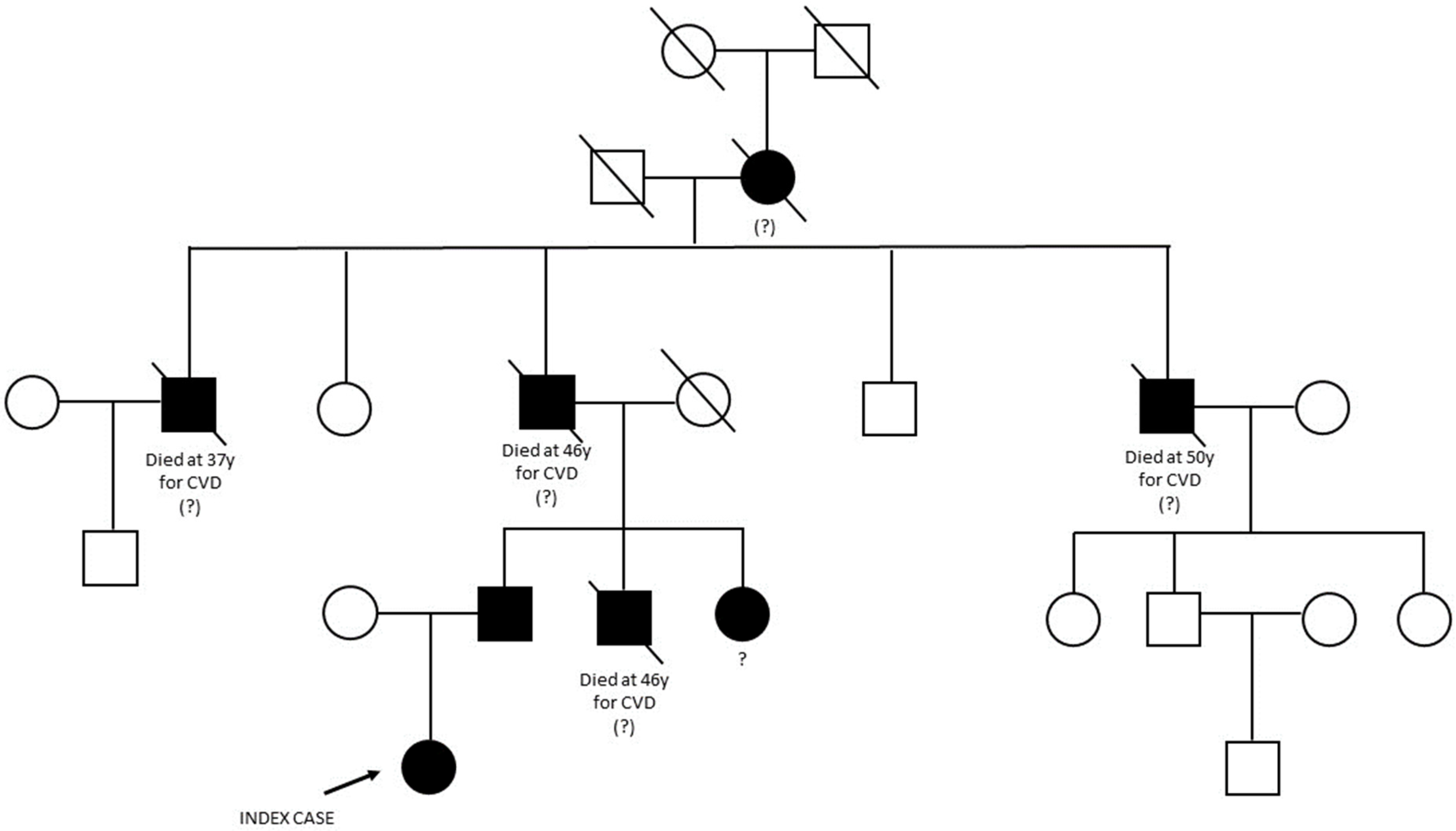
2.1. Case Presentation and Family History
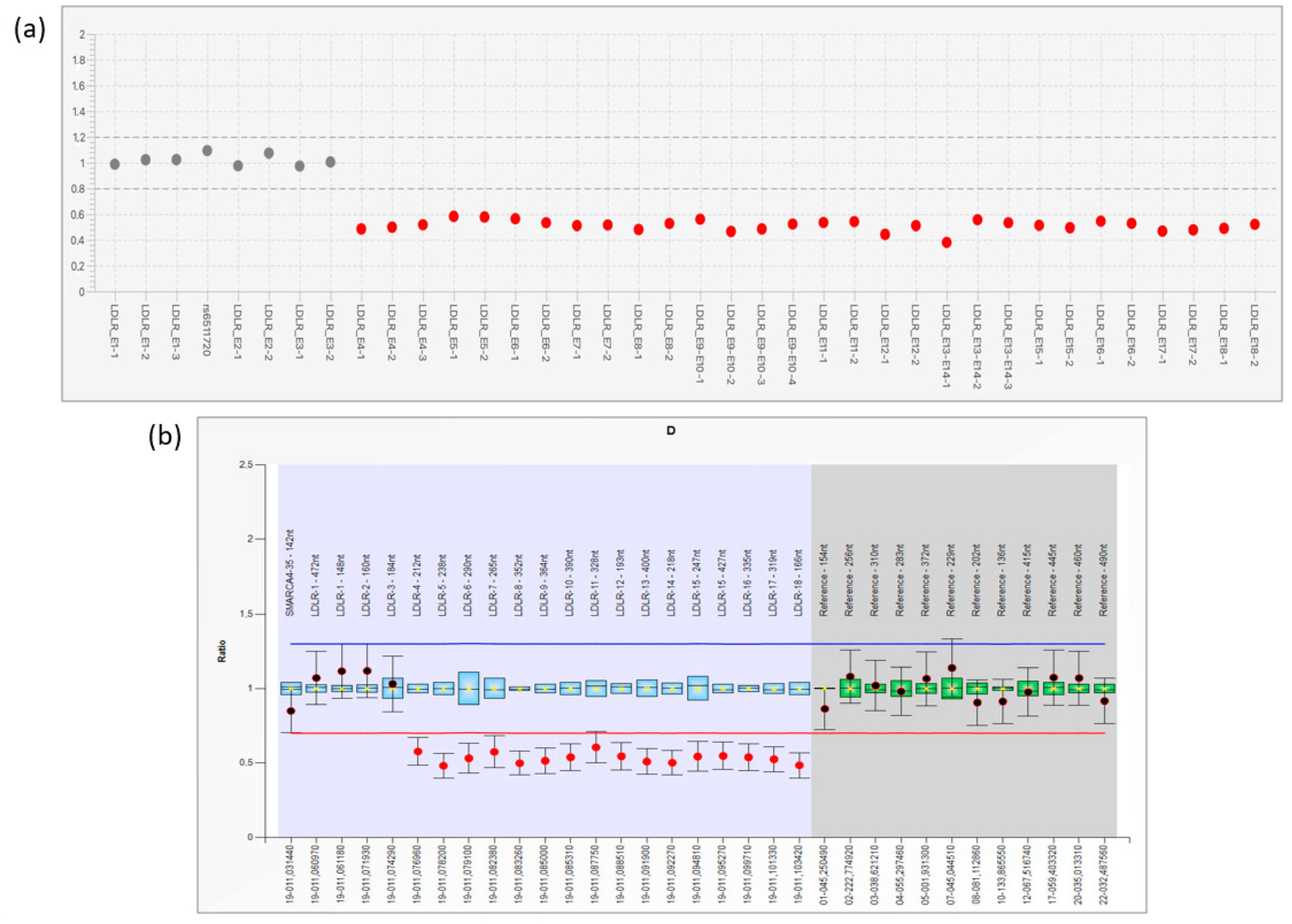
2.2. NGS and Large Genomic Rearrangement (LGR) Detection
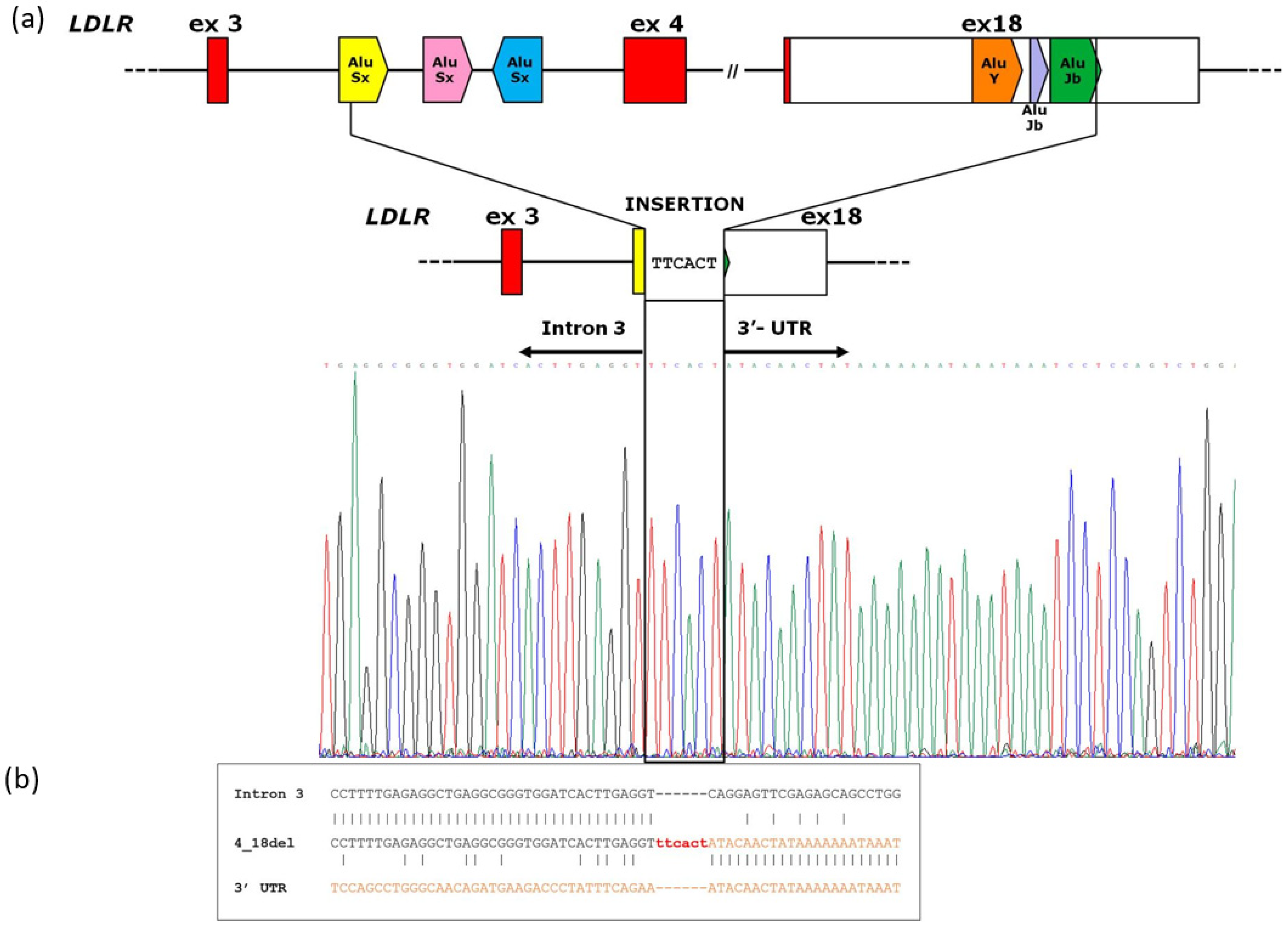
2.3. Analysis of Breakpoint Region
3. Results
3.1. NGS Analysis and LRG Detection
3.2. Analysis of Breakpoint Region
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Vaz, A.J.; Stevens, C.A.T.; Lyons, A.R.M.; Dharmayat, K.I.; Freiberger, T.; Hovingh, G.K.; Mata, P.; Raal, F.J.; Santos, R.D.; Soran, H.; et al. Global Perspective of Familial Hypercholesterolaemia: A Cross-Sectional Study from the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Lancet 2021, 398, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; de Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [Green Version]
- Haralambos, K.; Ashfield-Watt, P.; McDowell, I.F. Diagnostic scoring for familial hypercholesterolaemia in practice. Curr. Opin. Lipidol. 2016, 27, 367–374. [Google Scholar] [CrossRef]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrié, A.; Cuchel, M.; et al. Convened by the Familial Hypercholesterolemia Foundation. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef]
- Vallejo-Vaz, A.J.; De Marco, M.; Stevens, C.A.T.; Akram, A.; Freiberger, T.; Hovingh, G.K.; Kastelein, J.J.; Mata, P.; Raal, F.J.; Santos, R.D.; et al. Overview of the current status of familial hypercholesterolaemia care in over 60 countries—The EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Atherosclerosis 2018, 277, 234–255. [Google Scholar] [CrossRef] [Green Version]
- Moffa, S.; Onori, M.E.; De Paolis, E.; Ricciardi Tenore, C.; Perrucci, A.; Pontecorvi, A.; Giaccari, A.; Urbani, A.; Minucci, A. A novel low-density lipoprotein receptor variant in a Ukrainian patient: A case report and overview of the disease-causing low-density lipoprotein receptor variants associated to familial hypercholesterolemia. Mol. Biol. Rep. 2022, 49, 1623–1630. [Google Scholar] [CrossRef]
- Iacocca, M.A.; Hegele, R.A. Role of DNA Copy Number Variation in Dyslipidemias. Curr. Opin. Lipidol. 2018, 29, 125–132. [Google Scholar] [CrossRef]
- Amsellem, S.; Briffaut, D.; Carrié, A.; Rabés, J.P.; Girardet, J.P.; Fredenrich, A.; Moulin, P.; Krempf, M.; Reznik, Y.; Vialettes, B.; et al. Intronic Mutations Outside of Alu-Repeat-Rich Domains of the LDL Receptor Gene Are a Cause of Familial Hypercholesterolemia. Hum. Genet. 2002, 111, 501–510. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Banach, M.; Mancini, G.J.; Lepor, N.E.; Hanselman, J.C.; Zhao, X.; Leiter, L.A. Efficacy and safety of bempedoic acid added to ezetimibe in statin-intolerant patients with hypercholesterolemia: A randomized, placebo-controlled study. Atherosclerosis 2018, 277, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Burgess, L.J.; Ebenbichler, C.F.; Baum, S.J.; Stroes, E.S.; Ali, S.; Khilla, N.; Hamlin, R.; Pordy, R.; Dong, Y.; et al. Evinacumab in Patients with Refractory Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Defesche, J.C.; Lansberg, P.J.; Umans-Eckenhausen, M.A.W. Advanced method for the identification of patients with inherited hypercholesterolemia. Semin. Vasc. Med. 2004, 4, 59–65. [Google Scholar] [CrossRef]
- Moffa, S.; Mazzuccato, G.; De Bonis, M.; De Paolis, E.; Onori, M.E.; Pontecorvi, A.; Urbani, A.; Giaccari, A.; Capoluongo, E.; Minucci, A. Identification of two novel LDLR variants by Next Generation Sequencing. Ann. Ist Super. Sanita. 2020, 56, 122–127. [Google Scholar] [PubMed]
- RepeatMasker. Available online: https://www.repeatmasker.org (accessed on 11 May 2023).
- Südhof, T.C.; Goldstein, J.L.; Brown, M.S.; Russell, D.W. The LDL receptor gene: A mosaic of exons shared with different proteins. Science 1985, 228, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Gent, J.; Braakman, I. Low-density lipoprotein receptor structure and folding. Cell Mol. Life Sci. 2004, 61, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, H.H.; Russell, D.W.; Brown, M.S.; Goldstein, J.L. The LDL receptor locus in familial hypercholesterolemia: Mutational analysis of a membrane protein. Annu. Rev. Genet. 1990, 24, 133–170. [Google Scholar] [CrossRef]
- Goldmann, R.; Tichý, L.; Freiberger, T.; Zapletalová, P.; Letocha, O.; Soska, V.; Fajkus, J.; Fajkusová, L. Genomic characterization of large rearrangements of the LDLR gene in Czech patients with familial hypercholesterolemia. BMC Med. Genet. 2010, 11, 115. [Google Scholar] [CrossRef] [Green Version]
- Dron, J.S.; Wang, J.; McIntyre, A.D.; Iacocca, M.A.; Robinson, J.F.; Ban, M.R.; Cao, H.; Hegele, R.A. Six years’ experience with LipidSeq: Clinical and research learnings from a hybrid, targeted sequencing panel for dyslipidemias. BMC Med. Genom. 2020, 13, 2322. [Google Scholar] [CrossRef] [Green Version]
- Rutkowska, L.; Pinkier, I.; Sałacińska, K.; Kępczyński, Ł.; Salachna, D.; Lewek, J.; Banach, M.; Matusik, P.; Starostecka, E.; Lewiński, A.; et al. Identification of New Copy Number Variation and the Evaluation of a CNV Detection Tool for NGS Panel Data in Polish Familial Hypercholesterolemia Patients. Genes 2022, 13, 1424. [Google Scholar] [CrossRef]
- Iacocca, M.A.; Wang, J.; Dron, J.S.; Robinson, J.F.; McIntyre, A.D.; Cao, H.; Hegele, R.A. Use of next-generation sequencing to detect LDLR gene copy number variation in familial hypercholesterolemia. J. Lipid Res. 2017, 58, 2202–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Concolino, P.; Rizza, R.; Mignone, F.; Costella, A.; Guarino, D.; Carboni, I.; Capoluongo, E.; Santonocito, C.; Urbani, A.; Minucci, A. A comprehensive BRCA1/2 NGS pipeline for an immediate Copy Number Variation (CNV) detection in breast and ovarian cancer molecular diagnosis. Clin. Chim. Acta. 2018, 480, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Lazarte, J.; Berberich, A.J.; Wang, J.; Hegele, R.A. A cautionary tale: Is this APOB whole-gene duplication actually pathogenic? J. Clin. Lipidol. 2020, 14, 631–663. [Google Scholar] [CrossRef] [PubMed]
- Iacocca, M.A.; Wang, J.; Sarkar, S.; Dron, J.S.; Lagace, T.; McIntyre, A.D.; Lau, P.; Robinson, J.F.; Yang, P.; Knoll, J.H.; et al. Whole-Gene Duplication of PCSK9 as a Novel Genetic Mechanism for Severe Familial Hypercholesterolemia. Can. J. Cardiol. 2018, 34, 1316–1324. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Lipid Profile | Years | Normal Range | ||
|---|---|---|---|---|
| 1994 | 2019 | 2022 | ||
| Total Cholesterol (mg/dL) | 374 | 357 | 321 | 130–200 mg/dL |
| HDL | 93 | 68 | 66 | female > 45; male > 40 |
| Triglycerides (mg/dL) | 281 | 124 | 95 | 20–170 mg/dL |
| LDL (mg/dL) | 225 | 264 | 236 | <130 mg/dL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Concolino, P.; De Paolis, E.; Moffa, S.; Onori, M.E.; Soldovieri, L.; Ricciardi Tenore, C.; De Bonis, M.; Rabacchi, C.; Santonocito, C.; Rinelli, M.; et al. Identification and Molecular Characterization of a Novel Large-Scale Variant (Exons 4_18 Loss) in the LDLR Gene as a Cause of Familial Hypercholesterolaemia in an Italian Family. Genes 2023, 14, 1275. https://doi.org/10.3390/genes14061275
Concolino P, De Paolis E, Moffa S, Onori ME, Soldovieri L, Ricciardi Tenore C, De Bonis M, Rabacchi C, Santonocito C, Rinelli M, et al. Identification and Molecular Characterization of a Novel Large-Scale Variant (Exons 4_18 Loss) in the LDLR Gene as a Cause of Familial Hypercholesterolaemia in an Italian Family. Genes. 2023; 14(6):1275. https://doi.org/10.3390/genes14061275
Chicago/Turabian StyleConcolino, Paola, Elisa De Paolis, Simona Moffa, Maria Elisabetta Onori, Laura Soldovieri, Claudio Ricciardi Tenore, Maria De Bonis, Claudio Rabacchi, Concetta Santonocito, Martina Rinelli, and et al. 2023. "Identification and Molecular Characterization of a Novel Large-Scale Variant (Exons 4_18 Loss) in the LDLR Gene as a Cause of Familial Hypercholesterolaemia in an Italian Family" Genes 14, no. 6: 1275. https://doi.org/10.3390/genes14061275
APA StyleConcolino, P., De Paolis, E., Moffa, S., Onori, M. E., Soldovieri, L., Ricciardi Tenore, C., De Bonis, M., Rabacchi, C., Santonocito, C., Rinelli, M., Calandra, S., Giaccari, A., Urbani, A., & Minucci, A. (2023). Identification and Molecular Characterization of a Novel Large-Scale Variant (Exons 4_18 Loss) in the LDLR Gene as a Cause of Familial Hypercholesterolaemia in an Italian Family. Genes, 14(6), 1275. https://doi.org/10.3390/genes14061275