
Suppression of NANOG Expression Reduces Drug Resistance of Cancer Stem Cells in Glioblastoma



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Production of Lentivirus Containing shRNA
2.3. Lentiviral Transduction
2.4. Two-Step Quanitative Reverse Transcription-PCR
2.5. Cell Viability Assay
2.6. Cell Cycle Analysis
2.7. Statistical Analysis and Graphs
3. Results
3.1. Suppression of Stemness Genes NANOG and OCT4 Expressions by Short Hairpin RNA Interference (shRNA)
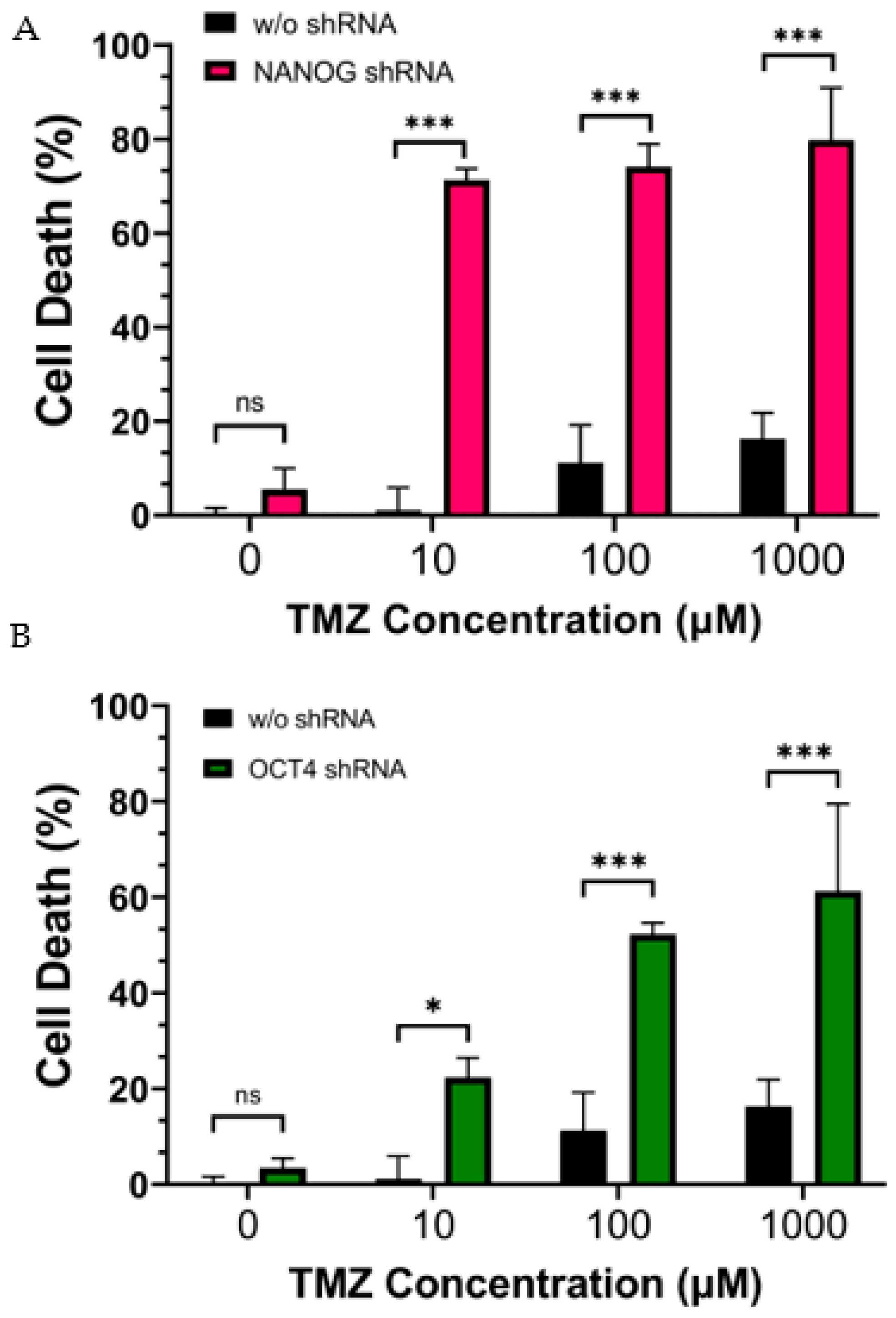
3.2. Reduction in Stemness Gene Expression Increased CSC Vulnerability against TMZ
3.3. Suppression of NANOG Did Not Affect MGMT Expression
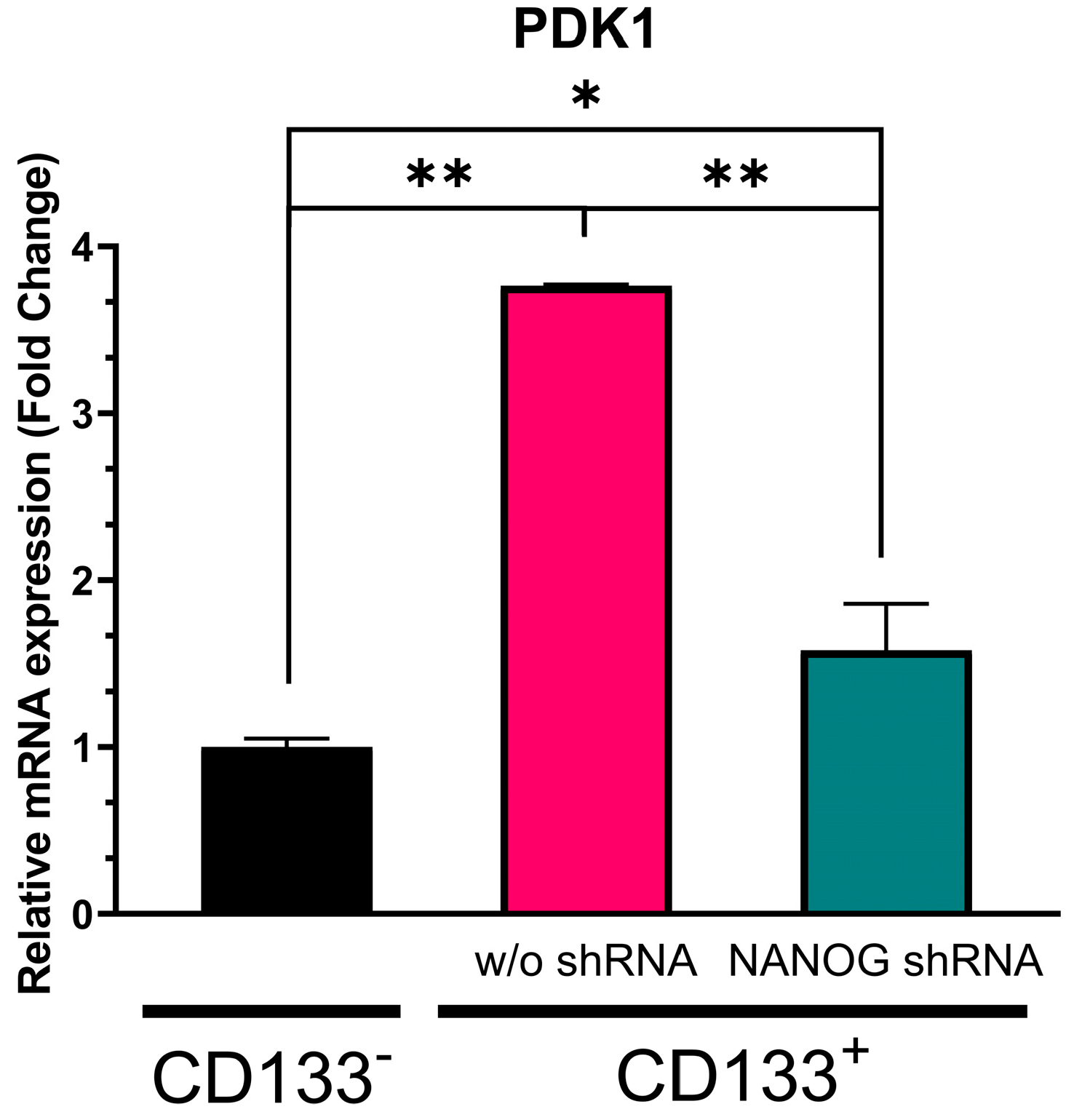
3.4. Effect of NANOG Suppression on PI3K/AKT Pathway
3.5. Suppression of NANOG Expression Caused G0–G1 Arrest of GSCs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3. [Google Scholar]
- Fernandes, C.; Costa, A.; Osorio, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Copyright: The Authors.: Brisbane (AU); Codon Publications: Singapore, 2017. [Google Scholar]
- Mann, J.; Ramakrishna, R.; Magge, R.; Wernicke, A.G. Advances in radiotherapy for glioblastoma. Front. Neurol. 2018, 8, 748. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Shackleton, M. Normal stem cells and cancer stem cells: Similar and different. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2010; pp. 85–92. [Google Scholar]
- Rossi, F.; Noren, H.; Jove, R.; Beljanski, V.; Grinnemo, K.-H. Differences and similarities between cancer and somatic stem cells: Therapeutic implications. Stem Cell Res. Ther. 2020, 11, 489. [Google Scholar] [CrossRef]
- Aponte, P.M.; Caicedo, A. Stemness in cancer: Stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Jaggupilli, A.; Elkord, E. Significance of CD44 and CD24 as cancer stem cell markers: An enduring ambiguity. Clin. Dev. Immunol. 2012, 2012, 708036. [Google Scholar] [CrossRef] [Green Version]
- Geng, S.; Guo, Y.; Wang, Q.; Li, L.; Wang, J. Cancer stem-like cells enriched with CD29 and CD44 markers exhibit molecular characteristics with epithelial–mesenchymal transition in squamous cell carcinoma. Arch. Dermatol. Res. 2013, 305, 35–47. [Google Scholar] [CrossRef]
- Li, Z. CD133: A stem cell biomarker and beyond. Exp. Hematol. Oncol. 2013, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Liang, X.; Zhan, Y.; Wang, Z.; Xu, J.; Qiu, Y.; Wang, J.; Cao, Y.; Le, V.-M.; Ly, H.-T. Targeting CD133 reverses drug-resistance via the AKT/NF-κB/MDR1 pathway in colorectal cancer. Br. J. Cancer 2020, 122, 1342–1353. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and Cancer stem cells. Cell Commun. Signal. 2021, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma-a comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Seymour, T.; Nowak, A.; Kakulas, F. Targeting aggressive cancer stem cells in glioblastoma. Front. Oncol. 2015, 5, 159. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Ortiz, R.; Perazzoli, G.; Cabeza, L.; Jiménez-Luna, C.; Luque, R.; Prados, J.; Melguizo, C. Temozolomide: An updated overview of resistance mechanisms, nanotechnology advances and clinical applications. Curr. Neuropharmacol. 2021, 19, 513–537. [Google Scholar]
- Ohba, S.; Yamashiro, K.; Hirose, Y. Inhibition of DNA repair in combination with temozolomide or dianhydrogalactiol overcomes temozolomide-resistant glioma cells. Cancers 2021, 13, 2570. [Google Scholar] [CrossRef]
- Vaidya, M.; Sreerama, S.; Gonzalez-Vega, M.; Smith, J.; Field, M.; Sugaya, K. Coculture with Neural Stem Cells May Shift the Transcription Profile of Glioblastoma Multiforme towards Cancer-Specific Stemness. Int. J. Mol. Sci. 2023, 24, 3242. [Google Scholar] [CrossRef]
- Oldrini, B.; Vaquero-Siguero, N.; Mu, Q.; Kroon, P.; Zhang, Y.; Galán-Ganga, M.; Bao, Z.; Wang, Z.; Liu, H.; Sa, J.K. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat. Commun. 2020, 11, 3883. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell. Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Shah, N.; Lin, B.; Sibenaller, Z.; Ryken, T.; Lee, H.; Yoon, J.G.; Rostad, S.; Foltz, G. Comprehensive analysis of MGMT promoter methylation: Correlation with MGMT expression and clinical response in GBM. PLoS ONE 2011, 6, e16146. [Google Scholar] [CrossRef]
- Steed, T.C.; Treiber, J.M.; Taha, B.; Engin, H.B.; Carter, H.; Patel, K.S.; Dale, A.M.; Carter, B.S.; Chen, C.C. Glioblastomas located in proximity to the subventricular zone (SVZ) exhibited enrichment of gene expression profiles associated with the cancer stem cell state. J. Neuro-Oncol. 2020, 148, 455–462. [Google Scholar] [CrossRef]
- Field, M.; Alvarez, A.; Bushnev, S.; Sugaya, K. Embryonic stem cell markers distinguishing cancer stem cells from normal human neuronal stem cell populations in malignant glioma patients. Clin. Neurosurg 2010, 57, 151–159. [Google Scholar]
- Rasti, A.; Mehrazma, M.; Madjd, Z.; Abolhasani, M.; Saeednejad Zanjani, L.; Asgari, M. Co-expression of cancer stem cell markers OCT4 and NANOG predicts poor prognosis in renal cell carcinomas. Sci. Rep. 2018, 8, 11739. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, M.; Bacchus, M.; Sugaya, K. Differential sequences of exosomal NANOG DNA as a potential diagnostic cancer marker. PLoS ONE 2018, 13, e0197782. [Google Scholar] [CrossRef]
- Yoon, C.; Lu, J.; Yi, B.C.; Chang, K.K.; Simon, M.C.; Ryeom, S.; Yoon, S.S. PI3K/Akt pathway and Nanog maintain cancer stem cells in sarcomas. Oncogenesis 2021, 10, 12. [Google Scholar] [CrossRef]
- Lu, C.-S.; Shieh, G.-S.; Wang, C.-T.; Su, B.-H.; Su, Y.-C.; Chen, Y.-C.; Su, W.-C.; Wu, P.; Yang, W.-H.; Shiau, A.-L. Chemotherapeutics-induced Oct4 expression contributes to drug resistance and tumor recurrence in bladder cancer. Oncotarget 2017, 8, 30844. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Xie, C.M.; Li, H.; Tan, M.; Chen, G.; Schiff, R.; Xiong, X.; Sun, Y. The FBXW2-MSX2-SOX2 axis regulates stem cell property and drug resistance of cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 20528–20538. [Google Scholar] [CrossRef] [Green Version]
- Song, W.-S.; Yang, Y.-P.; Huang, C.-S.; Lu, K.-H.; Liu, W.-H.; Wu, W.-W.; Lee, Y.-Y.; Lo, W.-L.; Lee, S.-D.; Chen, Y.-W. Sox2, a stemness gene, regulates tumor-initiating and drug-resistant properties in CD133-positive glioblastoma stem cells. J. Chin. Med. Assoc. 2016, 79, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Wang, B.; Wang, X.; Luo, Y.; Fan, W. NANOGP8 is the key regulator of stemness, EMT, Wnt pathway, chemoresistance, and other malignant phenotypes in gastric cancer cells. PLoS ONE 2018, 13, e0192436. [Google Scholar] [CrossRef] [Green Version]
- Jeter, C.R.; Badeaux, M.; Choy, G.; Chandra, D.; Patrawala, L.; Liu, C.; Calhoun-Davis, T.; Zaehres, H.; Daley, G.Q.; Tang, D.G. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells 2009, 27, 993–1005. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, A.; Wickremesekera, A.; Brasch, H.D.; Chibnall, A.M.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer stem cells in glioblastoma multiforme. Front. Surg. 2016, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Manni, W.; Min, W. Signaling pathways in the regulation of cancer stem cells and associated targeted therapy. MedComm 2022, 3, e176. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- Park, S.R.; Kim, S.R.; Hong, I.S.; Lee, H.Y. A Novel Therapeutic Approach for Colorectal Cancer Stem Cells: Blocking the PI3K/Akt Signaling Axis With Caffeic Acid. Front. Cell Dev. Biol. 2020, 8, 585987. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Allahverdi, A.; Arefian, E.; Soleimani, M.; Ai, J.; Nahanmoghaddam, N.; Yousefi-Ahmadipour, A.; Ebrahimi-Barough, S. MicroRNA-4731-5p delivered by AD-mesenchymal stem cells induces cell cycle arrest and apoptosis in glioblastoma. J. Cell. Physiol. 2020, 235, 8167–8175. [Google Scholar] [CrossRef]
- Juan-García, A.; Tolosa, J.; Juan, C.; Ruiz, M.-J. Cytotoxicity, genotoxicity and disturbance of cell cycle in HepG2 cells exposed to OTA and BEA: Single and combined actions. Toxins 2019, 11, 341. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Watt, F.M. Nanog maintains pluripotency of mouse embryonic stem cells by inhibiting NFkappaB and cooperating with Stat3. Nat. Cell Biol. 2008, 10, 194–201. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12, e8195. [Google Scholar] [CrossRef]
- Jakovlevs, A.; Vanags, A.; Gardovskis, J.; Strumfa, I. Molecular classification of diffuse gliomas. Pol. J. Pathol. 2019, 70, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, H.; Xu, S.; Liu, Z.; Cheng, Q. The adaptive transition of glioblastoma stem cells and its implications on treatments. Signal Transduct. Target. Ther. 2021, 6, 124. [Google Scholar] [CrossRef]
- Najafzadeh, B.; Asadzadeh, Z.; Motafakker Azad, R.; Mokhtarzadeh, A.; Baghbanzadeh, A.; Alemohammad, H.; Abdoli Shadbad, M.; Vasefifar, P.; Najafi, S.; Baradaran, B. The oncogenic potential of NANOG: An important cancer induction mediator. J. Cell. Physiol. 2021, 236, 2443–2458. [Google Scholar] [CrossRef]
- Hassn Mesrati, M.; Behrooz, A.B.; Abuhamad, A.Y.; Syahir, A. Understanding glioblastoma biomarkers: Knocking a mountain with a hammer. Cells 2020, 9, 1236. [Google Scholar] [CrossRef]
- Li, B.; McCrudden, C.M.; Yuen, H.F.; Xi, X.; Lyu, P.; Chan, K.W.; Zhang, S.D.; Kwok, H.F. CD133 in brain tumor: The prognostic factor. Oncotarget 2017, 8, 11144–11159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J.; et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, M.; Yan, G.; Ma, Q.; Yan, Z.; Wang, L.; Yang, K.; Guo, D. Nanog promotes stem-like traits of glioblastoma cells. Front. Biosci. 2021, 26, 552–565. [Google Scholar] [CrossRef]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. Embo J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.J.; Herlyn, M. The emerging roles of Oct4 in tumor-initiating cells. Am. J. Physiol. Cell Physiol. 2015, 309, C709–C718. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006, 38, 431–440. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Hsu, H.-S.; Chen, Y.-W.; Tsai, T.-H.; How, C.-K.; Wang, C.-Y.; Hung, S.-C.; Chang, Y.-L.; Tsai, M.-L.; Lee, Y.-Y. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS ONE 2008, 3, e2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilghman, J.; Wu, H.; Sang, Y.; Shi, X.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; Eberhart, C.G.; Laterra, J.; Ying, M. HMMR maintains the stemness and tumorigenicity of glioblastoma stem-like cells. Cancer Res. 2014, 74, 3168–3179. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.-K.; Lin, J.W.; Shih, J.-W.; Chuang, H.-Y.; Fong, I.-H.; Yeh, C.-T.; Lin, C.-M. Targeting BC200/miR218-5p signaling axis for overcoming temozolomide resistance and suppressing glioma stemness. Cells 2020, 9, 1859. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [Green Version]
- Langhans, J.; Schneele, L.; Trenkler, N.; von Bandemer, H.; Nonnenmacher, L.; Karpel-Massler, G.; Siegelin, M.D.; Zhou, S.; Halatsch, M.-E.; Debatin, K.-M. The effects of PI3K-mediated signalling on glioblastoma cell behaviour. Oncogenesis 2017, 6, 398. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.M.; Wu, G.L.; Huang, J.; Li, J.G.; Hao, C.; He, Q.M.; Chen, X.D.; Wang, G.X.; Tu, X.H. Low expression of PDK1 inhibits renal cell carcinoma cell proliferation, migration, invasion and epithelial mesenchymal transition through inhibition of the PI3K-PDK1-Akt pathway. Cell. Signal. 2019, 56, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bamodu, O.A.; Chang, H.L.; Ong, J.R.; Lee, W.H.; Yeh, C.T.; Tsai, J.T. Elevated PDK1 Expression Drives PI3K/AKT/MTOR Signaling Promotes Radiation-Resistant and Dedifferentiated Phenotype of Hepatocellular Carcinoma. Cells 2020, 9, 746. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; Flynn, D.C.; Zhang, Z.; Zhong, X.S.; Walker, V.; Liu, K.J.; Shi, X.; Jiang, B.H. G1 cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/mTOR/p70S6K1 signaling in human ovarian cancer cells. Am. J. Physiol. Cell Physiol. 2004, 287, C281–C291. [Google Scholar] [CrossRef]
- Guo, Q.; Xiong, Y.; Song, Y.; Hua, K.; Gao, S. ARHGAP17 suppresses tumor progression and up-regulates P21 and P27 expression via inhibiting PI3K/AKT signaling pathway in cervical cancer. Gene 2019, 692, 9–16. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| NANOG-F | 5′-GTCTTCTGCTGAGATGCCTCACA-3′ |
| NANOG-R | 5′-TCTGCTGGAGGCTGAGGTAT-3′ |
| OCT4-F | 5′-GGAAGGTATTCAGCCAAACGACCA-3′ |
| OCT4-R | 5′-CTCACTCGGTTCTCGATACTGGTT-3′ |
| MGMT-F | 5′-TTCACCATCCCGTTTTCCAG-3′ |
| MGMT-R | 5′-ATTGCCTCTCATTGCTCCTC-3′ |
| PDK1-F | 5′-TCGTCCTCCTCCTCACACTCCCT-3′ |
| PDK1-R | 5′-GCCTGCTTCTCCAACAACAACCTCTT-3′ |
| B-Actin-F | 5′-AGAGCTACGAGCTGCCTGAC-3′ |
| B-Actin-R | 5′-AGCACTGTGTTGGCGTACAG-3′ |
| Combination of TMZ and shRNA | CDI |
|---|---|
| TMZ 10 µM + shRNA NANOG | 0.25 |
| TMZ 100 µM + shRNA NANOG | 0.26 |
| TMZ 1000 µM + shRNA NANOG | 0.28 |
| TMZ 10 µM + shRNA OCT4 | 0.2 |
| TMZ 100 µM + shRNA OCT4 | 0.18 |
| TMZ 1000 µM + shRNA OCT4 | 0.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, J.; Field, M.; Sugaya, K. Suppression of NANOG Expression Reduces Drug Resistance of Cancer Stem Cells in Glioblastoma. Genes 2023, 14, 1276. https://doi.org/10.3390/genes14061276
Smith J, Field M, Sugaya K. Suppression of NANOG Expression Reduces Drug Resistance of Cancer Stem Cells in Glioblastoma. Genes. 2023; 14(6):1276. https://doi.org/10.3390/genes14061276
Chicago/Turabian StyleSmith, Jonhoi, Melvin Field, and Kiminobu Sugaya. 2023. "Suppression of NANOG Expression Reduces Drug Resistance of Cancer Stem Cells in Glioblastoma" Genes 14, no. 6: 1276. https://doi.org/10.3390/genes14061276
APA StyleSmith, J., Field, M., & Sugaya, K. (2023). Suppression of NANOG Expression Reduces Drug Resistance of Cancer Stem Cells in Glioblastoma. Genes, 14(6), 1276. https://doi.org/10.3390/genes14061276