
Large-Scale Whole Genome Sequence Analysis of >22,000 Subjects Provides no Evidence of FMR1 Premutation Allele Involvement in Autism Spectrum Disorder



Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples with WGS Data
2.2. Genotype Assessment of the CGG Trinucleotide Repeat at FMR1
2.3. PCR Validation of FMR1 Premutation Carriers and Their Pedigrees
2.4. Repeat Burden Statistical Testing
3. Results
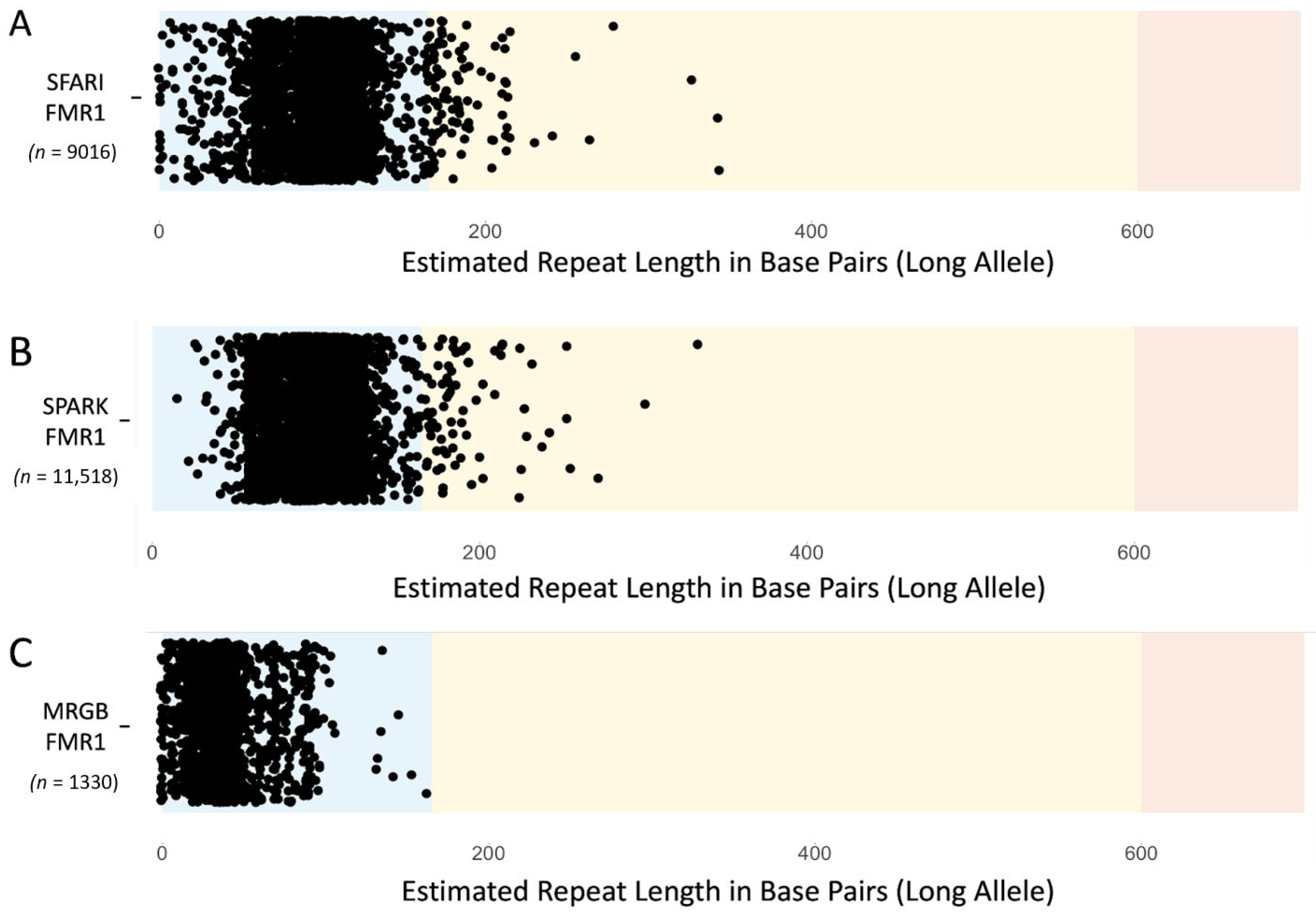
3.1. Validation of FMR1 Repeat Alleles Calculated by ExpansionHunter
3.2. Prevalence of Individuals with FMR1 Repeat Alleles
3.3. Burden of FMR1 Premutation Range Alleles on ASD in Males and Females
3.4. Parental Transmission of FMR1 Premutation Range Alleles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Garber, K.B.; Visootsak, J.; Warren, S.T. Fragile X syndrome. Eur. J. Hum. Genet. 2008, 16, 666–672. [Google Scholar] [CrossRef] [Green Version]
- Hall, D.A. In the Gray Zone in the Fragile X Gene: What are the Key Unanswered Clinical and Biological Questions? Tremor Other Hyperkinet Mov. 2014, 4, 208. [Google Scholar] [CrossRef]
- Nolin, S.L.; Glicksman, A.; Tortora, N.; Allen, E.; Macpherson, J.; Mila, M.; Vianna-Morgante, A.M.; Sherman, S.L.; Dobkin, C.; Latham, G.J.; et al. Expansions and contractions of the FMR1 CGG repeat in 5508 transmissions of normal, intermediate, and premutation alleles. Am. J. Med. Genet. A 2019, 179, 1148–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.E.; Berry-Kravis, E.; Hipp, H.; Todd, P.K. FMR1 Disorders. GeneReviews®. 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1384/ (accessed on 1 August 2022).
- Rousseau, F.; Rouillard, P.; Morel, M.L.; Khandjian, E.W.; Morgan, K. Prevalence of carriers of premutation-size alleles of the FMRI gene--and implications for the population genetics of the fragile X syndrome. Am. J. Hum. Genet. 1995, 57, 1006–1018. [Google Scholar] [PubMed]
- Hantash, F.M.; Goos, D.M.; Crossley, B.; Anderson, B.; Zhang, K.; Sun, W.; Strom, C.M. FMR1 premutation carrier frequency in patients undergoing routine population-based carrier screening: Insights into the prevalence of fragile X syndrome, fragile X-associated tremor/ataxia syndrome, and fragile X-associated primary ovarian insufficiency in the United States. Genet. Med. 2011, 13, 39–45. [Google Scholar]
- Seltzer, M.M.; Baker, M.W.; Hong, J.; Maenner, M.; Greenberg, J.; Mandel, D. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 589–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassone, F.; Iong, K.P.; Tong, T.-H.; Lo, J.; Gane, L.W.; Berry-Kravis, E.; Nguyen, D.; Mu, L.Y.; Laffin, J.; Bailey, D.B.; et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012, 4, 100. [Google Scholar] [CrossRef] [Green Version]
- Maenner, M.J.; Baker, M.W.; Broman, K.W.; Tian, J.; Barnes, J.K.; Atkins, A.; McPherson, E.; Hong, J.; Brilliant, M.H.; Mailick, M.R. FMR1 CGG expansions: Prevalence and sex ratios. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013, 162B, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Hagerman, R.; Au, J.; Hagerman, P. FMR1 premutation and full mutation molecular mechanisms related to autism. J. Neurodev. Disord. 2011, 3, 211–224. [Google Scholar] [CrossRef] [Green Version]
- Abbeduto, L.; McDuffie, A.; Thurman, A.J. The fragile X syndrome autism comorbidity: What do we really know? Front. Genet. 2014, 5, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movaghar, A.; Page, D.; Brilliant, M.; Baker, M.W.; Greenberg, J.; Hong, J.; DaWalt, L.S.; Saha, K.; Kuusisto, F.; Stewart, R.; et al. Data-driven phenotype discovery of FMR1 premutation carriers in a population-based sample. Sci. Adv. 2019, 5, eaaw7195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabal-Herrera, A.M.; Saldarriaga-Gil, W.; Salcedo-Arellano, M.J.; Hagerman, R.J. Fragile X associated neuropsychiatric disorders in a male without FXTAS. Intractable Rare Dis. Res. 2020, 9, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Aishworiya, R.; Protic, D.; Tang, S.J.; Schneider, A.; Tassone, F.; Hagerman, R. Fragile X-Associated Neuropsychiatric Disorders (FXAND) in Young Fragile X Premutation Carriers. Genes 2022, 13, 2399. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H. Genetics of autism spectrum disorders. Trends Cogn. Sci. 2011, 15, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The Heritability of Autism Spectrum Disorder. JAMA 2017, 318, 1182–1184. [Google Scholar] [CrossRef]
- Niu, M.; Han, Y.; Dy, A.B.C.; Du, J.; Jin, H.; Qin, J.; Zhang, J.; Li, Q.; Hagerman, R.J. Autism Symptoms in Fragile X Syndrome. J. Child Neurol. 2017, 32, 903–909. [Google Scholar] [CrossRef]
- Fernandez, B.A.; Scherer, S.W. Syndromic autism spectrum disorders: Moving from a clinically defined to a molecularly defined approach. Dialogues Clin. Neurosci. 2017, 19, 353–371. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Klein, S.D.; Nguyen, D.C.; Kim, A.; Signer, R.; Fox, M.; Dorrani, N.; Hendershot, A.; Mardach, R.; Suddath, R.; et al. Is it time to retire fragile X testing as a first-tier test for developmental delay, intellectual disability, and autism spectrum disorder? Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 1380. [Google Scholar] [CrossRef] [Green Version]
- Fischbach, G.D.; Lord, C. The Simons Simplex Collection: A Resource for dentification of Autism Genetic Risk Factors. Neuron 2010, 68, 192–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SPARK Consortium. SPARK: A US Cohort of 50,000 Families to Accelerate Autism Research. Neuron 2018, 97, 488–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolzhenko, E.; van Vugt, J.J.; Shaw, R.J.; Bekritsky, M.A.; van Blitterswijk, M.; Narzisi, G.; Ajay, S.S.; Rajan, V.; Lajoie, B.R.; Johnson, N.H.; et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017, 27, 1895–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolzhenko, E.; Deshpande, V.; Schlesinger, F.; Krusche, P.; Petrovski, R.; Chen, S.; Emig-Agius, D.; Gross, A.; Narzisi, G.; Bowman, B.; et al. ExpansionHunter: A sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 2019, 35, 4754–4756. [Google Scholar] [CrossRef] [Green Version]
- Weisburd, B.; VanNoy, G.; Watts, N. The Addition of Short Tandem Repeat Calls to gnomAD. Available online: https://gnomad.broadinstitute.org/news/2022-01-the-addition-of-short-tandem-repeat-calls-to-gnomad/ (accessed on 24 August 2022).
- Matoba, N.; Liang, D.; Sun, H.; Aygün, N.; McAfee, J.C.; Davis, J.E.; Raffield, L.M.; Qian, H.; Piven, J.; Li, Y.; et al. Common genetic risk variants identified in the SPARK cohort support DDHD2 as a candidate risk gene for autism. Transl. Psychiatry 2020, 10, 265. [Google Scholar] [CrossRef]
- Wilfert, A.B.; Turner, T.N.; Murali, S.C.; Hsieh, P.; Sulovari, A.; Wang, T.; Coe, B.P.; Guo, H.; Hoekzema, K.; Bakken, T.E.; et al. Recent ultra-rare inherited variants implicate new autism candidate risk genes. Nat. Genet. 2021, 53, 1125–1134. [Google Scholar] [CrossRef]
- Pinese, M.; Lacaze, P.; Rath, E.M.; Stone, A.; Brion, M.-J.; Ameur, A.; Nagpal, S.; Puttick, C.; Husson, S.; Degrave, D.; et al. The Medical Genome Reference Bank contains whole genome and phenotype data of 2570 healthy elderly. Nat Commun. 2020, 11, 435. [Google Scholar] [CrossRef] [Green Version]
- Rice, J.A. Mathematical Statistics and Data Analysis; Cengage Learning: Boston, MA, USA, 2006. [Google Scholar]
- Curtis, D. Use of siblings as controls in case-control association studies. Ann. Hum. Genet. 1997, 61, 319–333. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Kidd, S.A.; Andrews, H.F.; Budimirovic, D.B.; Esler, A.; Haas-Givler, B.; Stackhouse, T.; Riley, C.; Peacock, G.; Sherman, S.L.; et al. Autism Spectrum Disorder in Fragile X Syndrome: Cooccurring Conditions and Current Treatment. Pediatrics 2017, 139, S194–S206. [Google Scholar] [CrossRef] [Green Version]
- Sherman, S.L.; Hunter, J.E. Epidemiology of Fragile X Syndrome. In Fragile X Syndrome; Academic Press: Cambridge, MA, USA, 2017; pp. 57–76. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | Family ID | Family Member | PCR Determined Length Genotype | ExpansionHunter Determined Repeat Length Genotype (with Off-Target Reads) | ExpansionHunter Determined Repeat Length Genotype (FMR1 Reference Region) |
|---|---|---|---|---|---|
| SSC02135 | 11372 | Father | Y/23 | Y/23 | Y/23 |
| SSC02130 | 11372 | Mother | 29/48 | 29/70 | 29/70 |
| SSC02128 | 11372 | Male Proband | Y/48 | Y/46 | Y/46 |
| SSC02136 | 11372 | Male Sibling | Y/48 | Y/57 | Y/51 |
| SSC02349 | 11676 | Father | Y/27 | Y/281 | Y/27 |
| SSC02338 | 11676 | Mother | 29/30 | 29/30 | 29/30 |
| SSC02330 | 11676 | Female Proband | 27/30 | 27/30 | 27/30 |
| SSC02350 | 11676 | Female Sibling | 27/29 | 27/29 | 27/29 |
| SSC07297 | 13390 | Father | Y/30 | Y/30 | Y/30 |
| SSC07289 | 13390 | Mother | 30/33 | 30/33 | 30/33 |
| SSC07282 | 13390 | Male Proband | Y/33 | Y/157 | Y/71 |
| SSC07298 | 13390 | Female Sibling | 30/30 | 30/30 | 30/30 |
| SSC12025 | 14489 | Father | Y/30 | Y/30 | Y/30 |
| SSC12020 | 14489 | Mother | 39/46 | 39/92 | 38/68 |
| SSC12016 | 14489 | Male Proband | Y/39 | Y/128 | Y/71 |
| SSC12026 | 14489 | Male Sibling | Y/39 | Y/168 | Y/72 |
| Family Member | Processed Samples | Samples with FMR1 Genotype | FMR1 Premutation Carriers | FMR1 Premutation Non-Carriers | Percentage of FMR1 Premutation Carriers |
|---|---|---|---|---|---|
| Father | 2364 | 2362 | 22 | 2340 | 0.93% |
| Mother | 2369 | 2365 | 42 | 2323 | 1.78% |
| Male Proband | 2060 | 2055 | 13 | 2042 | 0.63% |
| Male Sibling | 906 | 905 | 8 | 897 | 0.88% |
| Female Proband | 321 | 321 | 6 | 315 | 1.87% |
| Female Sibling | 1011 | 1008 | 11 | 997 | 1.09% |
| Total | 9031 | 9016 | 102 | 8914 | - |
| Family Member | Processed Samples | Samples with FMR1 Genotype | FMR1 Premutation Carriers | FMR1 Premutation Non-Carriers | Percentage of FMR1 Premutation Carriers |
|---|---|---|---|---|---|
| Father | 3075 | 3067 | 19 | 3048 | 0.62% |
| Mother | 3078 | 3077 | 22 | 3055 | 0.71% |
| Male Proband | 2509 | 2497 | 16 | 2481 | 0.64% |
| Male Sibling | 1133 | 1127 | 4 | 1123 | 0.35% |
| Female Proband | 614 | 614 | 7 | 607 | 1.14% |
| Female Sibling | 1136 | 1136 | 10 | 1126 | 0.88% |
| Total | 11545 | 11518 | 78 | 11446 | - |
| Parent to Offspring Transmission/De Novo Expansion in Offspring | Allele Transmitted | Count | Percentage of Offspring in FMR1 Premutation Families |
|---|---|---|---|
| Mother to Male Proband | Premutation Range Allele | 10 | 7.09% |
| Mother to Male Sibling | Premutation Range Allele | 7 | 4.96% |
| Mother to Female Proband | Premutation Range Allele | 3 | 2.12% |
| Mother to Female Sibling | Premutation Range Allele | 6 | 4.26% |
| Father to Male Proband | Premutation Range Allele | 0 | 0.00% |
| Father to Male Sibling | Premutation Range Allele | 0 | 0.00% |
| Father to Female Proband | Premutation Range Allele | 2 | 1.42% |
| Father to Female Sibling | Premutation Range Allele | 4 | 2.83% |
| Mother to Male Proband | Normal Range Allele | 19 | 13.48% |
| Mother to Male Sibling | Normal Range Allele | 14 | 9.92% |
| Mother to Female Proband | Normal Range Allele | 2 | 1.42% |
| Mother to Female Sibling | Normal Range Allele | 7 | 4.96% |
| Father to Male Proband | Normal Range Allele | 0 | 0.00% |
| Father to Male Sibling | Normal Range Allele | 0 | 0.00% |
| Father to Female Proband | Normal Range Allele | 0 | 0.00% |
| Father to Female Sibling | Normal Range Allele | 0 | 0.00% |
| Male Proband | de novo | 3 | 2.12% |
| Male Sibling | de novo | 1 | 0.71% |
| Female Proband | de novo | 0 | 0.00% |
| Female Sibling | de novo | 1 | 0.71% |
| Parent to Offspring Transmission/De Novo Expansion in Offspring | Allele Transmitted | Count | Percentage of Offspring in FMR1 Premutation Families |
|---|---|---|---|
| Mother to Male Proband | Premutation Range Allele | 15 | 15.15% |
| Mother to Male Sibling | Premutation Range Allele | 4 | 4.04% |
| Mother to Female Proband | Premutation Range Allele | 3 | 3.03% |
| Mother to Female Sibling | Premutation Range Allele | 4 | 4.04% |
| Father to Male Proband | Premutation Range Allele | 0 | 0.00% |
| Father to Male Sibling | Premutation Range Allele | 0 | 0.00% |
| Father to Female Proband | Premutation Range Allele | 3 | 3.03% |
| Father to Female Sibling | Premutation Range Allele | 5 | 5.05% |
| Mother to Male Proband | Normal Range Allele | 11 | 11.11% |
| Mother to Male Sibling | Normal Range Allele | 6 | 6.06% |
| Mother to Female Proband | Normal Range Allele | 2 | 2.02% |
| Mother to Female Sibling | Normal Range Allele | 7 | 7.07% |
| Father to Male Proband | Normal Range Allele | 0 | 0.00% |
| Father to Male Sibling | Normal Range Allele | 0 | 0.00% |
| Father to Female Proband | Normal Range Allele | 0 | 0.00% |
| Father to Female Sibling | Normal Range Allele | 0 | 0.00% |
| Male Proband | de novo | 1 | 0.01% |
| Male Sibling | de novo | 0 | 0.00% |
| Female Proband | de novo | 1 | 0.01% |
| Female Sibling | de novo | 1 | 0.01% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chubick, A.; Wang, E.; Au, C.; Grody, W.W.; Ophoff, R.A. Large-Scale Whole Genome Sequence Analysis of >22,000 Subjects Provides no Evidence of FMR1 Premutation Allele Involvement in Autism Spectrum Disorder. Genes 2023, 14, 1518. https://doi.org/10.3390/genes14081518
Chubick A, Wang E, Au C, Grody WW, Ophoff RA. Large-Scale Whole Genome Sequence Analysis of >22,000 Subjects Provides no Evidence of FMR1 Premutation Allele Involvement in Autism Spectrum Disorder. Genes. 2023; 14(8):1518. https://doi.org/10.3390/genes14081518
Chicago/Turabian StyleChubick, Alex, Evan Wang, Cora Au, Wayne W. Grody, and Roel A. Ophoff. 2023. "Large-Scale Whole Genome Sequence Analysis of >22,000 Subjects Provides no Evidence of FMR1 Premutation Allele Involvement in Autism Spectrum Disorder" Genes 14, no. 8: 1518. https://doi.org/10.3390/genes14081518
APA StyleChubick, A., Wang, E., Au, C., Grody, W. W., & Ophoff, R. A. (2023). Large-Scale Whole Genome Sequence Analysis of >22,000 Subjects Provides no Evidence of FMR1 Premutation Allele Involvement in Autism Spectrum Disorder. Genes, 14(8), 1518. https://doi.org/10.3390/genes14081518