
Multiple Sclerosis Heritability Estimation on Sardinian Ascertained Extended Families Using Bayesian Liability Threshold Model

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
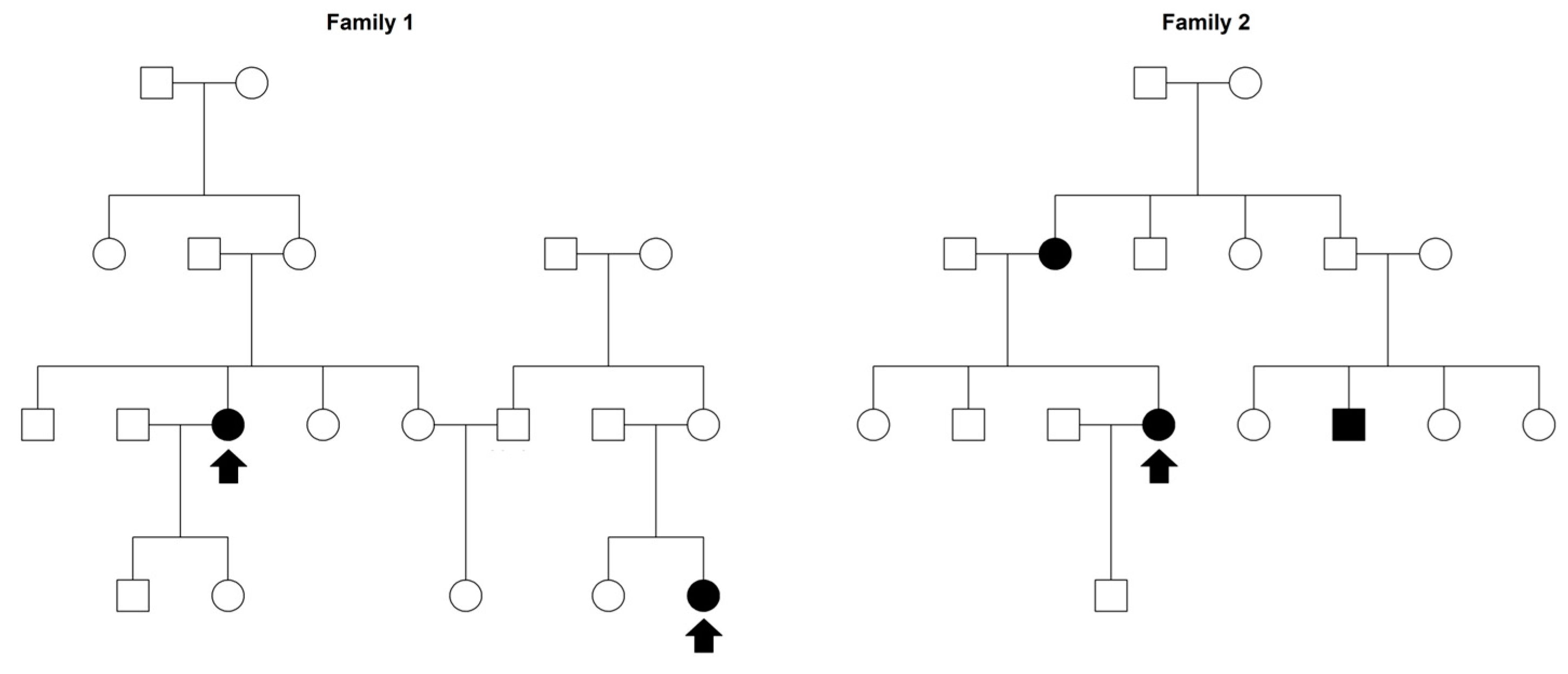
2.1. Sardinian Families Ascertainment
2.2. Statistical Analysis
2.2.1. Model Specification
2.2.2. Implementing Bayesian-LTMH
3. Results and Discussion
3.1. Sample Description
3.2. Bayesian-LTMH Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Visscher, P.M.; Hill, W.G.; Wray, N.R. Heritability in the genomics era—Concepts and misconceptions. Nat. Rev. Genet. 2008, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Kempthorne, O. The correlation between relatives on the supposition of mendelian inheritance. Am. J. Hum. Genet. 1968, 20, 402. [Google Scholar]
- Egeland, J. Heritability and Etiology: Heritability estimates can provide causally relevant information. Pers. Individ. Dif. 2023, 200, 111896. [Google Scholar] [CrossRef]
- Athanasiadis, G.; Speed, D.; Andersen, M.K.; Appel, E.V.R.; Grarup, N.; Brandslund, I.; Jørgensen, M.E.; Larsen, C.V.L.; Bjerregaard, P.; Hansen, T.; et al. Estimating narrow-sense heritability using family data from admixed populations. Heredity 2020, 124, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, R.; Spicer, R.; Muthukrishna, M. Cultural evolution of genetic heritability. Behav. Brain Sci. 2022, 45, e152. [Google Scholar] [CrossRef]
- Pearson, C.H. Is heritability explanatorily useful? Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 2007, 38, 270–288. [Google Scholar] [CrossRef]
- Bourrat, P. Heritability, causal influence and locality. Synthese 2021, 198, 6689–6715. [Google Scholar] [CrossRef]
- Tal, O. From heritability to probability. Biol. Philos. 2009, 24, 81–105. [Google Scholar] [CrossRef]
- Milo, R.; Kahana, E. Multiple sclerosis: Geoepidemiology, genetics and the environment. Autoimmun. Rev. 2010, 9, A387–A394. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Schriml, L.M.; Lichenstein, R.; Bisordi, K.; Bearer, C.; Baron, J.A.; Greene, C. Modeling the enigma of complex disease etiology. J. Transl. Med. 2023, 21, 148. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses andCell-Based Therapy. Cell J. 2017, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Fazia, T.; Marzanati, D.; Carotenuto, A.L.; Beecham, A.; Hadjixenofontos, A.; McCauley, J.L.; Saddi, V.; Piras, M.; Bernardinelli, L.; Gentilini, D. Homozygosity Haplotype and Whole-Exome Sequencing Analysis to Identify Potentially Functional Rare Variants Involved in Multiple Sclerosis among Sardinian Families. Curr. Issues Mol. Biol. 2021, 43, 1778–1793. [Google Scholar] [CrossRef] [PubMed]
- Fazia, T.; Baldrighi, G.N.; Nova, A.; Bernardinelli, L. A systematic review of Mendelian randomization studies on multiple sclerosis. Eur. J. Neurosci. 2023. [Google Scholar] [CrossRef] [PubMed]
- Waubant, E.; Lucas, R.; Mowry, E.; Graves, J.; Olsson, T.; Alfredsson, L.; Langer-Gould, A. Environmental and genetic risk factors for MS: An integrated review. Ann. Clin. Transl. Neurol. 2019, 6, 1905–1922. [Google Scholar] [CrossRef]
- Amato, M.P.; Derfuss, T.; Hemmer, B.; Liblau, R.; Montalban, X.; Soelberg Sørensen, P.; Miller, D.H.; Alfredsson, L.; Aloisi, F.; Ascherio, A.; et al. Environmental modifiable risk factors for multiple sclerosis: Report from the 2016 ECTRIMS focused workshop. Mult. Scler. 2018, 24, 590–603. [Google Scholar] [CrossRef]
- Patsopoulos, N.A.; Baranzini, S.E.; Santaniello, A.; Shoostari, P.; Cotsapas, C.; Wong, G.; Beecham, A.H.; James, T.; Replogle, J.; Vlachos, I.S.; et al. Multiple Sclerosis Genomic Map implicates peripheral immune cells & microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Mitrovič, M.; Patsopoulos, N.A.; Beecham, A.H.; Dankowski, T.; Goris, A.; Dubois, B.; D’hooghe, M.B.; Lemmens, R.; Van Damme, P.; Søndergaard, H.B.; et al. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell 2020, 180, 403. [Google Scholar] [CrossRef]
- Fazia, T.; Pastorino, R.; Foco, L.; Han, L.; Abney, M.; Beecham, A.; Hadjixenofontos, A.; Guo, H.; Gentilini, D.; Papachristou, C.; et al. Investigating multiple sclerosis genetic susceptibility on the founder population of east-central Sardinia via association and linkage analysis of immune-related loci. Mult. Scler. 2018, 24, 1815–1824. [Google Scholar] [CrossRef]
- Fagnani, C.; Neale, M.C.; Nisticò, L.; Stazi, M.A.; Ricigliano, V.A.; Buscarinu, M.C.; Salvetti, M.; Ristori, G. Twin studies in multiple sclerosis: A meta-estimation of heritability and environmentality. Mult. Scler. 2015, 21, 1404–1413. [Google Scholar] [CrossRef]
- Ristori, G.; Cannoni, S.; Stazi, M.A.; Vanacore, N.; Cotichini, R.; Alfò, M.; Pugliatti, M.; Sotgiu, S.; Solaro, C.; Bomprezzi, R.; et al. Multiple Sclerosis in Twins from Continental Italy and Sardinia: A Nationwide Study. Ann. Neurol. 2005, 59, 27–34. [Google Scholar] [CrossRef]
- Kruuk, L.E.B.; Hadfield, J.D. How to separate genetic and environmental causes of similarity between relatives. J. Evol. Biol. 2007, 20, 1890–1903. [Google Scholar] [CrossRef] [PubMed]
- Dick, D.M. Shared Environment. Encycl. Stat. Behav. Sci. 2005, 4, 1828–1830. [Google Scholar] [CrossRef]
- Kendler, K.S.; Ohlsson, H.; Lichtenstein, P.; Sundquist, J.; Sundquist, K. The Nature of the Shared Environment. Behav. Genet. 2019, 49, 1–10. [Google Scholar] [CrossRef]
- Pittner, K.; Bakermans-Kranenburg, M.J.; Alink, L.R.A.; Buisman, R.S.M.; van den Berg, L.J.M.; van den Block, L.H.C.D.; Voorthuis, A.; Elzinga, B.M.; Lindenberg, J.; Tollenaar, M.S.; et al. Estimating the Heritability of Experiencing Child Maltreatment in an Extended Family Design. Child Maltreat. 2020, 25, 289. [Google Scholar] [CrossRef]
- De Villemereuil, P.; Gimenez, O.; Doligez, B. Comparing parent–offspring regression with frequentist and Bayesian animal models to estimate heritability in wild populations: A simulation study for Gaussian and binary traits. Methods Ecol. Evol. 2013, 4, 260–275. [Google Scholar] [CrossRef]
- Park, S.; Lee, S.; Lee, Y.; Herold, C.; Hooli, B.; Mullin, K.; Park, T.; Park, C.; Bertram, L.; Lange, C.; et al. Adjusting heterogeneous ascertainment bias for genetic association analysis with extended families. BMC Med. Genet. 2015, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Macgregor, A.J. Twin studies and the heritability of MS: A conclusion. Mult. Scler. 2009, 15, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kwak, S.H.; Won, S. Heritability estimation of dichotomous phenotypes using a liability threshold model on ascertained family-based samples. Genet. Epidemiol. 2019, 43, 761–775. [Google Scholar] [CrossRef]
- Louis, T.A. Finding the Observed Information Matrix When Using the EM Algorithm. J. R. Stat. Soc. Ser. B 1982, 44, 226–233. [Google Scholar] [CrossRef]
- Xu, C.; Baines, P.D.; Wang, J.L. Standard error estimation using the EM algorithm for the joint modeling of survival and longitudinal data. Biostatistics 2014, 15, 731. [Google Scholar] [CrossRef] [PubMed]
- Sofer, T. Confidence intervals for heritability via Haseman-Elston regression. Stat. Appl. Genet. Mol. Biol. 2017, 16, 259. [Google Scholar] [CrossRef] [PubMed]
- Granieri, E.; Casetta, I.; Govoni, V.; Tola, M.R.; Marchi, D.; Murgia, S.B.; Ticca, A.; Pugliatti, M.; Murgia, B.; Rosati, G. The increasing incidence and prevalence of MS in a Sardinian province. Neurology 2000, 55, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, S.; Pugliatti, M.; Sotgiu, A.; Sanna, A.; Rosati, G. Review: Does the “Hygiene Hypothesis” Provide an Explanation for the High Prevalence of Multiple Sclerosis in Sardinia? Autoimmunity 2009, 36, 257–260. [Google Scholar] [CrossRef]
- Sotgiu, S.; Pugliatti, M.; Sanna, A.; Sotgiu, A.; Castigli, P.; Solinas, G.; Dolei, A.; Serra, C.; Bonetti, B.; Rosati, G. Multiple sclerosis complexity in selected populations: The challenge of Sardinia, insular Italy. Eur. J. Neurol. 2002, 9, 329–341. [Google Scholar] [CrossRef]
- Matveeva, O.; Bogie, J.F.J.; Hendriks, J.J.A.; Linker, R.A.; Haghikia, A.; Kleinewietfeld, M. Western lifestyle and immunopathology of multiple sclerosis. Ann. N. Y. Acad. Sci. 2018, 1417, 71. [Google Scholar] [CrossRef]
- Tognotti, E. Program to Eradicate Malaria in Sardinia, 1946–1950. Emerg. Infect. Dis. 2009, 15, 1460. [Google Scholar] [CrossRef]
- Riedl, B.; Beckmann, T.; Neundõrfer, B.; Handwerker, H.O.; Birklein, F. Multiple sclerosis epidemiology in Sardinia: Evidence for a true increasing risk. Acta Neurol. Scand. 2001, 103, 20–26. [Google Scholar] [CrossRef]
- Casetta, I.; Granieri, E.; Marchi, D.; Murgia, S.B.; Tola, M.R.; Ticca, A.; Lauria, G.; Govoni, V.; Murgia, B.; Pugliatti, M. An epidemiological study of multiple sclerosis in central Sardinia, Italy. Acta Neurol. Scand. 1998, 98, 391–394. [Google Scholar] [CrossRef]
- Poser, C.M.; Paty, D.W.; Scheinberg, L.; McDonald, W.I.; Davis, F.A.; Ebers, G.C.; Johnson, K.P.; Sibley, W.A.; Silberberg, D.H.; Tourtellotte, W.W. New diagnostic criteria for multiple sclerosis: Guidelines for research protocols. Ann. Neurol. 1983, 13, 227–231. [Google Scholar] [CrossRef]
- Hujoel, M.L.A.; Gazal, S.; Loh, P.R.; Patterson, N.; Price, A.L. Liability threshold modeling of case–control status and family history of disease increases association power. Nat. Genet. 2020, 52, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Montomoli, C.; Allemani, C.; Solinas, G.; Motta, G.; Bernardinelli, L.; Clemente, S.; Murgia, B.S.; Ticca, A.F.; Musu, L.; Piras, M.L.; et al. An ecologic study of geographical variation in multiple sclerosis risk in central Sardinia, Italy. Neuroepidemiology 2002, 21, 187–193. [Google Scholar] [CrossRef]
- Urru, S.A.M.; Antonelli, A.; Sechi, G.M. Prevalence of multiple sclerosis in Sardinia: A systematic cross-sectional multi-source survey. Mult. Scler. J. 2020, 26, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R. Concepts and Misconceptions about the Polygenic Additive Model Applied to Disease. Hum. Hered. 2015, 80, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Ebers, G.C.; Sadovnick, A.D.; Dyment, D.A.; Yee, I.M.L.; Willer, C.J.; Risch, N. Parent-of-origin effect in multiple sclerosis: Observations in half-siblings. Lancet 2004, 363, 1773–1774. [Google Scholar] [CrossRef]
- Hoppenbrouwers, I.A.; Liu, F.; Aulchenko, Y.S.; Ebers, G.C.; Oostra, B.A.; Van Duijn, C.M.; Hintzen, R.Q. Maternal transmission of multiple sclerosis in a dutch population. Arch. Neurol. 2008, 65, 345–348. [Google Scholar] [CrossRef]
- Benchek, P.H.; Morris, N.J. How meaningful are heritability estimates of liability? Hum. Genet. 2013, 132, 1351–1360. [Google Scholar] [CrossRef]
- Gjessing, H.K.; Lie, R.T. Biometrical modelling in genetics: Are complex traits too complex? Stat. Methods Med. Res. 2008, 17, 75–96. [Google Scholar] [CrossRef]
- de Villemereuil, P.; Morrissey, M.B.; Nakagawa, S.; Schielzeth, H. Fixed-effect variance and the estimation of repeatabilities and heritabilities: Issues and solutions. J. Evol. Biol. 2018, 31, 621–632. [Google Scholar] [CrossRef]
- Nakagawa, S.; Schielzeth, H. A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol. Evol. 2013, 4, 133–142. [Google Scholar] [CrossRef]
- Almasy, L.; Towne, B.; Peterson, C.; Blangero, J. Detecting genotype x age interaction. Genet. Epidemiol. 2001, 21 (Suppl. 1), S819–S824. [Google Scholar] [CrossRef]
- Poveda, A.; Chen, Y.; Brändström, A.; Engberg, E.; Hallmans, G.; Johansson, I.; Renström, F.; Kurbasic, A.; Franks, P.W. The heritable basis of gene-environment interactions in cardiometabolic traits. Diabetologia 2017, 60, 442–452. [Google Scholar] [CrossRef]
- Paap, R. What are the advantages of MCMC based inference in latent variable models? Stat. Neerl. 2002, 56, 2–22. [Google Scholar] [CrossRef]
- Dunson, D.B. Commentary: Practical Advantages of Bayesian Analysis of Epidemiologic Data. Am. J. Epidemiol. 2001, 153, 1222–1226. [Google Scholar] [CrossRef]
- Luengo, D.; Martino, L.; Bugallo, M.; Elvira, V.; Särkkä, S. A survey of Monte Carlo methods for parameter estimation. EURASIP J. Adv. Signal Process. 2020, 2020, 25. [Google Scholar] [CrossRef]
- Tang, Y. Beyond EM: A faster Bayesian linear regression algorithm without matrix inversions. Neurocomputing 2020, 378, 435–440. [Google Scholar] [CrossRef]
- Hamra, G.; MacLehose, R.; Richardson, D. Markov Chain Monte Carlo: An introduction for epidemiologists. Int. J. Epidemiol. 2013, 42, 627. [Google Scholar] [CrossRef] [PubMed]
- Elston, R.C.; Olson, J.M.; Palmer, L. Biostatistical Genetics and Genetic Epidemiology; John Wiley & Sons: Hoboken, NJ, USA, 2002; p. 831. [Google Scholar]
- Carpenter, B.; Gelman, A.; Hoffman, M.D.; Lee, D.; Goodrich, B.; Betancourt, M.; Brubaker, M.A.; Guo, J.; Li, P.; Riddell, A. Stan: A Probabilistic Programming Language. J. Stat. Softw. 2017, 76, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Betancourt, M. A Conceptual Introduction to Hamiltonian Monte Carlo. arXiv 2018, arXiv:1701.02434. [Google Scholar]
- Jonah Gabry and Rok Cesnovar cmdstanr: R Interface to “CmdStan”. 2021.
- Sotgiu, S.; Angius, A.; Embry, A.; Rosati, G.; Musumeci, S. Hygiene hypothesis: Innate immunity, malaria and multiple sclerosis. Med. Hypotheses 2008, 70, 819–825. [Google Scholar] [CrossRef]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Handunnetthi, L.; Giovannoni, G.; Ramagopalan, S.V. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS ONE 2010, 5, e12496. [Google Scholar] [CrossRef] [PubMed]
- Mameli, G.; Cossu, D.; Cocco, E.; Masala, S.; Frau, J.; Marrosu, M.G.; Sechi, L.A. EBNA-1 IgG titers in Sardinian multiple sclerosis patients and controls. J. Neuroimmunol. 2013, 264, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Alfredsson, L.; Olsson, T. Lifestyle and Environmental Factors in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a028944. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L.; Lünemann, J.D. The initiation and prevention of multiple sclerosis. Nat. Rev. Neurol. 2012, 8, 602–612. [Google Scholar] [CrossRef]
- Puthenparampil, M.; Perini, P.; Bergamaschi, R.; Capobianco, M.; Filippi, M.; Gallo, P. Multiple sclerosis epidemiological trends in Italy highlight the environmental risk factors. J. Neurol. 2022, 269, 1817–1824. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Family | Individuals N (%) 1 | Probands N | Females N (%) 2 | MS Cases N (%) 2 |
|---|---|---|---|---|
| 1 | 65 (8%) | 6 | 37 (57%) | 6 (9%) |
| 2 | 35 (4%) | 4 | 20 (57%) | 5 (14%) |
| 3 | 70 (9%) | 7 | 45 (64%) | 9 (13%) |
| 4 | 66 (8%) | 8 | 37 (56%) | 10 (15%) |
| 5 | 12 (2%) | 2 | 6 (50%) | 3 (25%) |
| 6 | 16 (2%) | 2 | 7 (44%) | 2 (13%) |
| 7 | 43 (5%) | 5 | 24 (56%) | 5 (12%) |
| 8 | 33 (4%) | 5 | 16 (48%) | 6 (18%) |
| 9 | 17 (2%) | 2 | 10 (59%) | 2 (12%) |
| 10 | 20 (3%) | 2 | 13 (65%) | 3 (15%) |
| 11 | 15 (2%) | 1 | 8 (53%) | 3 (20%) |
| 12 | 33 (4%) | 5 | 17 (52%) | 6 (18%) |
| 13 | 17 (2%) | 2 | 11 (65%) | 3 (18%) |
| 14 | 51 (6%) | 6 | 24 (47%) | 12 (24%) |
| 15 | 25 (3%) | 3 | 16 (64%) | 3 (12%) |
| 16 | 44 (6%) | 5 | 24 (55%) | 8 (18%) |
| 17 | 19 (2%) | 2 | 12 (63%) | 2 (11%) |
| 18 | 16 (2%) | 2 | 8 (50%) | 2 (13%) |
| 19 | 22 (3%) | 3 | 13 (59%) | 3 (14%) |
| 20 | 27 (3%) | 2 | 16 (59%) | 2 (7%) |
| 21 | 28 (4%) | 1 | 13 (46%) | 2 (7%) |
| 22 | 16 (2%) | 2 | 7 (44%) | 4 (25%) |
| 23 | 7 (1%) | 1 | 3 (43%) | 1 (14%) |
| 24 | 93 (12%) | 11 | 48 (52%) | 16 (17%) |
| Total | 790 | 89 | 435 (55%) | 118 (15%) |
| MS Course ° | N (%) | Females (%) | Age MS Onset Mean (SD) | Year MS Onset Mean (SD) |
|---|---|---|---|---|
| RRMS | 58 (49%) | 41 (71%) | 28.45 (9.49) | 1990 (10.09) |
| SPMS | 27 (23%) | 14 (52%) | 28.89 (8.87) | 1983 (9.64) |
| PPMS | 1 (1%) | 1 (100%) | 45.00 | 1995 |
| Unknown | 32 (27%) | 20 (63%) | N/A | N/A |
| Total | 118 | 76 (64%) | 28.64 (9.06) * | 1988 (10.88) * |
| Kinship Relationship | N (%) * |
|---|---|
| First degree | 20 (8%) |
| Parent–offspring | 9 |
| Mother | 6 |
| Father | 3 |
| Sibling | 13 |
| Second degree | 9 (4%) |
| Uncle/aunt–nephew/niece | 8 |
| Grandparent–grandchild | 1 |
| Third degree | 16 (7%) |
| Cousins | 15 |
| Grand-grandparent–grand-grandchild | 1 |
| Fourth degree | 17 (7%) |
| Over the fourth degree | 176 (74%) |
| Total | 238 |
| Parameter | Median | SD 1 | HPD 95% CI 1 |
|---|---|---|---|
| h2 | 0.033 | 0.028 | 0.000, 0.094 |
| c2Sibs | 0.033 | 0.016 | 0.007, 0.067 |
| c2Mother–Sibs | 0.012 | 0.012 | 0.000, 0.039 |
| c2Father–Sibs | 0.013 | 0.013 | 0.000, 0.040 |
| c2Spouses | 0.014 | 0.017 | 0.000, 0.051 |
| c2Total | 0.080 | 0.037 | 0.021, 0.158 |
| e2 | 0.168 | 0.036 | 0.094, 0.233 |
| τ2βSEX,YR | 0.712 | 0.020 | 0.673, 0.749 |
| τ2βSEX | 0.009 | 0.008 | 0.000, 0.027 |
| τ2βYR | 0.686 | 0.024 | 0.637, 0.731 |
| 2cov°βSEX,YR | 0.015 | 0.007 | 0.003, 0.028 |
| βSEX(Females vs. Males) | 0.355 | 0.157 | 0.057, 0.679 |
| βYR(≥1946 vs. <1946) | 3.173 | 0.155 | 2.869, 3.477 |
| Year of Birth < 1946 | Year of Birth ≥ 1946 | |||||
|---|---|---|---|---|---|---|
| Parameter | Median | SD 1 | 95% HPD CI 1 | Median | SD 1 | 95% HPD CI 1 |
| h2 | 0.090 | 0.100 | 0.000, 0.312 | 0.818 | 0.068 | 0.679, 0.937 |
| c2Sibs | 0.223 | 0.100 | 0.055, 0.433 | 0.045 | 0.030 | 0.004, 0.109 |
| c2Mother–Sibs | 0.061 | 0.058 | 0.000, 0.185 | 0.013 | 0.016 | 0.000, 0.050 |
| c2Father–Sibs | 0.049 | 0.051 | 0.000, 0.163 | 0.014 | 0.017 | 0.000, 0.054 |
| c2Spouses | 0.085 | 0.083 | 0.000, 0.297 | 0.019 | 0.026 | 0.000, 0.078 |
| c2Total | 0.477 | 0.142 | 0.199, 0.750 | 0.105 | 0.056 | 0.019, 0.222 |
| e2 | 0.086 | 0.083 | 0.000, 0.265 | 0.021 | 0.025 | 0.000, 0.078 |
| τ2βSEX,YR | N/A 1 | N/A 1 | N/A 1 | 0.042 | 0.032 | 0.000, 0.109 |
| τ2βSEX | 0.304 | 0.112 | 0.079, 0.506 | 0.005 | 0.013 | 0.000, 0.035 |
| τ2βYR | N/A 1 | N/A 1 | N/A 1 | 0.032 | 0.030 | 0.001, 0.095 |
| 2cov°βSEX,YR | N/A 1 | N/A 1 | N/A 1 | 0.000 | 0.001 | −0.001, 0.001 |
| βSEX(Females vs. Males) | 1.322 | 0.368 | 0.586, 2.023 | 0.104 | 0.177 | −0.246, 0.448 |
| βYR(10 years increase) | N/A 1 | N/A 1 | N/A 1 | 0.186 | 0.089 | 0.012, 0.362 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nova, A.; Fazia, T.; Saddi, V.; Piras, M.; Bernardinelli, L. Multiple Sclerosis Heritability Estimation on Sardinian Ascertained Extended Families Using Bayesian Liability Threshold Model. Genes 2023, 14, 1579. https://doi.org/10.3390/genes14081579
Nova A, Fazia T, Saddi V, Piras M, Bernardinelli L. Multiple Sclerosis Heritability Estimation on Sardinian Ascertained Extended Families Using Bayesian Liability Threshold Model. Genes. 2023; 14(8):1579. https://doi.org/10.3390/genes14081579
Chicago/Turabian StyleNova, Andrea, Teresa Fazia, Valeria Saddi, Marialuisa Piras, and Luisa Bernardinelli. 2023. "Multiple Sclerosis Heritability Estimation on Sardinian Ascertained Extended Families Using Bayesian Liability Threshold Model" Genes 14, no. 8: 1579. https://doi.org/10.3390/genes14081579
APA StyleNova, A., Fazia, T., Saddi, V., Piras, M., & Bernardinelli, L. (2023). Multiple Sclerosis Heritability Estimation on Sardinian Ascertained Extended Families Using Bayesian Liability Threshold Model. Genes, 14(8), 1579. https://doi.org/10.3390/genes14081579