
Neuroinflammatory Pathways in the ALS-FTD Continuum: A Focus on Genetic Variants

 , , ,
, , ,  and
and {kind=link}
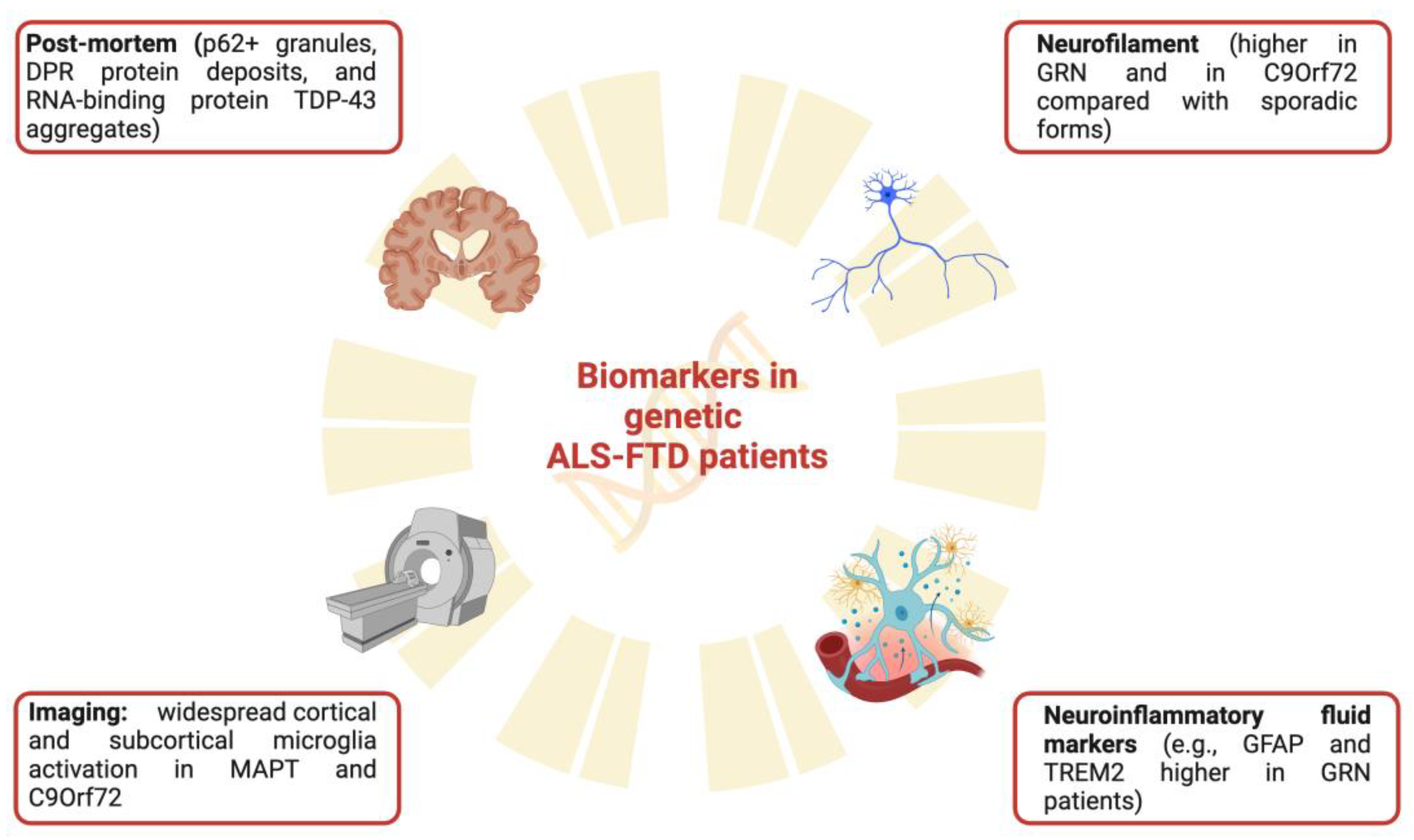{kind=link}
Abstract
:1. Introduction
2. Genetic in the ALS-FTD Continuum
3. Neuroinflammation in Neurodegenerative Diseases: Players and Pathways
4. Neuroinflammatory Pathways in the Genetic ALS-FTD Continuum
4.1. Post-Mortem and Preclinical Evidence
4.2. In Vivo and in Human Studies
4.2.1. Fluid Biomarkers
4.2.2. Neuroimaging Biomarkers
5. Autoimmune Disorders and Systemic Inflammation in Genetic ALS/FTD
6. Therapeutic Implications
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic Lateral Sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Mora, G.; Moglia, C.; Manera, U.; Canosa, A.; Cammarosano, S.; Ilardi, A.; Bertuzzo, D.; Bersano, E.; Cugnasco, P. Secular Trends of Amyotrophic Lateral Sclerosis: The Piemonte and Valle d’Aosta Register. JAMA Neurol. 2017, 74, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Tondo, G.; De Marchi, F. From Biomarkers to Precision Medicine in Neurodegenerative Diseases: Where Are We? J. Clin. Med. 2022, 11, 4515. [Google Scholar] [CrossRef] [PubMed]
- Vignaroli, F.; Mele, A.; Tondo, G.; De Giorgis, V.; Manfredi, M.; Comi, C.; Mazzini, L.; De Marchi, F. The Need for Biomarkers in the ALS–FTD Spectrum: A Clinical Point of View on the Role of Proteomics. Proteomes 2023, 11, 1. [Google Scholar] [CrossRef]
- Bang, J.; Spina, S.; Miller, B.L. Non-Alzheimer’s Dementia 1: Frontotemporal Dementia. Lancet 2015, 386, 1672. [Google Scholar] [CrossRef] [PubMed]
- Olney, N.T.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Neurol. Clin. 2017, 35, 339–374. [Google Scholar] [CrossRef]
- Tondo, G.; De Marchi, F.; Terazzi, E.; Sacchetti, M.; Cantello, R. Frontotemporal Dementia Presenting as Gambling Disorder: When a Psychiatric Condition Is the Clue to a Neurodegenerative Disease. Cogn. Behav. Neurol. 2017, 30, 62–67. [Google Scholar] [CrossRef]
- Rascovsky, K.; Grossman, M. Clinical Diagnostic Criteria and Classification Controversies in Frontotemporal Lobar Degeneration. Int. Rev. Psychiatry 2013, 25, 145–158. [Google Scholar] [CrossRef]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; Van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U. Sensitivity of Revised Diagnostic Criteria for the Behavioural Variant of Frontotemporal Dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- Liljegren, M.; Naasan, G.; Temlett, J.; Perry, D.C.; Rankin, K.P.; Merrilees, J.; Grinberg, L.T.; Seeley, W.W.; Englund, E.; Miller, B.L. Criminal Behavior in Frontotemporal Dementia and Alzheimer Disease. JAMA Neurol. 2015, 72, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Benussi, A.; Padovani, A.; Borroni, B. Phenotypic Heterogeneity of Monogenic Frontotemporal Dementia. Front. Aging Neurosci. 2015, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Burrell, J.R.; Halliday, G.M.; Kril, J.J.; Ittner, L.M.; Götz, J.; Kiernan, M.C.; Hodges, J.R. The Frontotemporal Dementia-Motor Neuron Disease Continuum. Lancet 2016, 388, 919–931. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Tondo, G.; Sarnelli, M.F.; Corrado, L.; Solara, V.; D’Alfonso, S.; Cantello, R.; Mazzini, L. A Case of Progressive Non-Fluent Aphasia as Onset of Amyotrophic Lateral Sclerosis with Frontotemporal Dementia. Int. J. Neurosci. 2019, 129, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Olszewska, D.A.; Lonergan, R.; Fallon, E.M.; Lynch, T. Genetics of Frontotemporal Dementia. Curr. Neurol. Neurosci. Rep. 2016, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Greaves, C.V.; Rohrer, J.D. An Update on Genetic Frontotemporal Dementia. J. Neurol. 2019, 266, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; Zucchi, E.; Martinelli, I.; Van der Most, L.; Gianferrari, G.; Moglia, C.; Manera, U.; Solero, L.; Vasta, R.; Canosa, A. Factors Predicting Disease Progression in C9ORF72 ALS Patients. J. Neurol. 2023, 270, 877–890. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and Impaired Regeneration of Gray Matter Oligodendrocytes in Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2013, 16, 571. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Boillée, S.; Velde, C.V.; Cleveland, D.W. ALS: A Disease of Motor Neurons and Their Nonneuronal Neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Ferrari, D.; Andjus, P.R.; Buzanska, L.; Cantello, R.; De Marchi, F.; Gelati, M.; Giniatullin, R.; Glover, J.C.; Grilli, M.; et al. Advances in Stem Cell Therapy for Amyotrophic Lateral Sclerosis. Expert. Opin. Biol. Ther. 2018, 18, 865–881. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Franjkic, T.; Schito, P.; Russo, T.; Nimac, J.; Chami, A.A.; Mele, A.; Vidatic, L.; Kriz, J.; Julien, J.-P. Emerging Trends in the Field of Inflammation and Proteinopathy in ALS/FTD Spectrum Disorder. Biomedicines 2023, 11, 1599. [Google Scholar] [CrossRef]
- Guerreiro, R.; Brás, J.; Hardy, J. SnapShot: Genetics of ALS and FTD. Cell 2015, 160, 798. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59. [Google Scholar] [CrossRef] [PubMed]
- Bali, T.; Self, W.; Liu, J.; Siddique, T.; Wang, L.H.; Bird, T.D.; Ratti, E.; Atassi, N.; Boylan, K.B.; Glass, J.D. Defining SOD1 ALS Natural History to Guide Therapeutic Clinical Trial Design. J. Neurol. Neurosurg. Psychiatry 2017, 88, 99–105. [Google Scholar] [CrossRef]
- Grassano, M.; Calvo, A.; Moglia, C.; Sbaiz, L.; Brunetti, M.; Barberis, M.; Casale, F.; Manera, U.; Vasta, R.; Canosa, A. Systematic Evaluation of Genetic Mutations in ALS: A Population-Based Study. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1190–1193. [Google Scholar] [CrossRef]
- Riva, N.; Pozzi, L.; Russo, T.; Pipitone, G.B.; Schito, P.; Domi, T.; Agosta, F.; Quattrini, A.; Carrera, P.; Filippi, M. NEK1 Variants in a Cohort of Italian Patients with Amyotrophic Lateral Sclerosis. Front. Neurosci. 2022, 16, 833051. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel Genes Associated with Amyotrophic Lateral Sclerosis: Diagnostic and Clinical Implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Van Swieten, J.C.; Heutink, P. Mutations in Progranulin (GRN) within the Spectrum of Clinical and Pathological Phenotypes of Frontotemporal Dementia. Lancet Neurol. 2008, 7, 965–974. [Google Scholar] [CrossRef]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited Review: Frontotemporal Dementia Caused by Microtubule-associated Protein Tau Gene (MAPT) Mutations: A Chameleon for Neuropathology and Neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zeng, P.; Fang, Y.; Zhang, T.; Tian, Q. Progranulin in Neurodegenerative Dementia. J. Neurochem. 2021, 158, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.J.; Logan, T.; DeVos, S.L.; Di Paolo, G. Lysosomal Functions of Progranulin and Implications for Treatment of Frontotemporal Dementia. Trends Cell Biol. 2022, 33, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Cruts, M.; Van Broeckhoven, C. The Role of Tau (MAPT) in Frontotemporal Dementia and Related Tauopathies. Hum. Mutat. 2004, 24, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT Mutations, Tauopathy, and Mechanisms of Neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, E.; De Marchi, F.; Contaldi, E.; Dianzani, U.; Cantello, R.; Mazzini, L.; Comi, C. The Role of Tau beyond Alzheimer’s Disease: A Narrative Review. Biomedicines 2022, 10, 760. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Kapogiannis, D.; D Huey, E.; Momeni, P. FTD and ALS: A Tale of Two Diseases. Curr. Alzheimer Res. 2011, 8, 273–294. [Google Scholar] [CrossRef] [PubMed]
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F. TARDBP Mutations in Individuals with Sporadic and Familial Amyotrophic Lateral Sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Cui, R.; Tuo, M.; Li, P.; Zhou, C. Association between TBK1 Mutations and Risk of Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Spectrum: A Meta-Analysis. Neurol. Sci. 2018, 39, 811–820. [Google Scholar] [CrossRef]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS Mutations Associated with Amyotrophic Lateral Sclerosis: Summary and Update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Zhang, Y.-J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.-F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.P. Novel Mutations in TARDBP (TDP-43) in Patients with Familial Amyotrophic Lateral Sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; Van Swieten, J.C.; Myllykangas, L. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Vatsavayai, S.C.; Nana, A.L.; Yokoyama, J.S.; Seeley, W.W. C9orf72-FTD/ALS Pathogenesis: Evidence from Human Neuropathological Studies. Acta Neuropathol. 2019, 137, 1–26. [Google Scholar] [CrossRef]
- Smeyers, J.; Banchi, E.-G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.K.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, Implicated in Amytrophic Lateral Sclerosis and Frontotemporal Dementia, Regulates Endosomal Trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef]
- Gendron, T.F.; Petrucelli, L. Disease Mechanisms of C9ORF72 Repeat Expansions. Cold Spring Harb. Perspect. Med. 2018, 8, a024224. [Google Scholar] [CrossRef]
- Le Ber, I.; Camuzat, A.; Guerreiro, R.; Bouya-Ahmed, K.; Bras, J.; Nicolas, G.; Gabelle, A.; Didic, M.; De Septenville, A.; Millecamps, S. SQSTM1 Mutations in French Patients with Frontotemporal Dementia or Frontotemporal Dementia with Amyotrophic Lateral Sclerosis. JAMA Neurol. 2013, 70, 1403–1410. [Google Scholar]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P. Haploinsufficiency of TBK1 Causes Familial ALS and Fronto-Temporal Dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Gijselinck, I.; Van Mossevelde, S.; van der Zee, J.; Sieben, A.; Philtjens, S.; Heeman, B.; Engelborghs, S.; Vandenbulcke, M.; De Baets, G.; Bäumer, V.; et al. Loss of TBK1 Is a Frequent Cause of Frontotemporal Dementia in a Belgian Cohort. Neurology 2015, 85, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Herhaus, L. TBK1 (TANK-Binding Kinase 1)-Mediated Regulation of Autophagy in Health and Disease. Matrix Biol. 2021, 100–101, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Koppers, M.; van Blitterswijk, M.M.; Vlam, L.; Rowicka, P.A.; van Vught, P.W.J.; Groen, E.J.N.; Spliet, W.G.M.; Engelen-Lee, J.; Schelhaas, H.J.; de Visser, M. VCP Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2012, 33, 837.e7–837.e13. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.-S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-Containing Protein (VCP) Is Required for Autophagy and Is Disrupted in VCP Disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H. Mutations of Optineurin in Amyotrophic Lateral Sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Pottier, C.; Bieniek, K.F.; Finch, N.; van de Vorst, M.; Baker, M.; Perkersen, R.; Brown, P.; Ravenscroft, T.; van Blitterswijk, M.; Nicholson, A.M. Whole-Genome Sequencing Reveals Important Role for TBK1 and OPTN Mutations in Frontotemporal Lobar Degeneration without Motor Neuron Disease. Acta Neuropathol. 2015, 130, 77–92. [Google Scholar] [CrossRef]
- Shen, W.-C.; Li, H.-Y.; Chen, G.-C.; Chern, Y.; Tu, P. Mutations in the Ubiquitin-Binding Domain of OPTN/Optineurin Interfere with Autophagy-Mediated Degradation of Misfolded Proteins by a Dominant-Negative Mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef]
- Lehmer, C.; Schludi, M.H.; Ransom, L.; Greiling, J.; Junghänel, M.; Exner, N.; Riemenschneider, H.; van der Zee, J.; Van Broeckhoven, C.; Weydt, P.; et al. A Novel CHCHD10 Mutation Implicates a Mia40-Dependent Mitochondrial Import Deficit in ALS. EMBO Mol. Med. 2018, 10, e8558. [Google Scholar] [CrossRef]
- Ronchi, D.; Riboldi, G.; Del Bo, R.; Ticozzi, N.; Scarlato, M.; Galimberti, D.; Corti, S.; Silani, V.; Bresolin, N.; Comi, G. Pietro CHCHD10 Mutations in Italian Patients with Sporadic Amyotrophic Lateral Sclerosis. Brain 2015, 138, e372. [Google Scholar] [CrossRef]
- Chaussenot, A.; Le Ber, I.; Ait-El-Mkadem, S.; Camuzat, A.; de Septenville, A.; Bannwarth, S.; Genin, E.C.; Serre, V.; Augé, G.; Brice, A. Screening of CHCHD10 in a French Cohort Confirms the Involvement of This Gene in Frontotemporal Dementia with Amyotrophic Lateral Sclerosis Patients. Neurobiol. Aging 2014, 35, 2884.e1–2884.e4. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Bandopadhyay, R.; Singh, P.K.; Mishra, P.S.; Sharma, N.; Khurana, N. Neuroinflammation in Neurological Disorders: Pharmacotherapeutic Targets from Bench to Bedside. Metab. Brain Dis. 2021, 36, 1591–1626. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in Neurodegenerative Disorders: The Roles of Microglia and Astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Comi, C.; Tondo, G. Insights into the Protective Role of Immunity in Neurodegenerative Disease. Neural Regen. Res. 2017, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS Neurodegenerative Diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Peferoen, L.; Kipp, M.; van der Valk, P.; van Noort, J.M.; Amor, S. Oligodendrocyte-microglia Cross-talk in the Central Nervous System. Immunology 2014, 141, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate Immune Activation in Neurodegenerative Disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Oksanen, M.; Lehtonen, S.; Jaronen, M.; Goldsteins, G.; Hämäläinen, R.H.; Koistinaho, J. Astrocyte Alterations in Neurodegenerative Pathologies and Their Modeling in Human Induced Pluripotent Stem Cell Platforms. Cell. Mol. Life Sci. 2019, 76, 2739–2760. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Huang, Y.; Bao, T.; Liu, C.; Liu, X.; Chen, X. The Role of Th17 Cells/IL-17A in AD, PD, ALS and the Strategic Therapy Targeting on IL-17A. J. Neuroinflammation 2022, 19, 98. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Winner, B.; Prots, I. The Trojan Horse-Neuroinflammatory Impact of T Cells in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic Reactions in Amyotrophic Lateral Sclerosis Brain and Spinal Cord Tissue. Am. J. Pathol. 1992, 140, 691. [Google Scholar] [PubMed]
- Engelhardt, J.I.; Tajti, J.; Appel, S.H. Lymphocytic Infiltrates in the Spinal Cord in Amyotrophic Lateral Sclerosis. Arch. Neurol. 1993, 50, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Hewitt, C.; Highley, J.R.; Brockington, A.; Milano, A.; Man, S.; Martindale, J.; Hartley, J.; Walsh, T.; Gelsthorpe, C. Clinico-Pathological Features in Amyotrophic Lateral Sclerosis with Expansions in C9ORF72. Brain 2012, 135, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.M.; Livesey, M.R.; McDade, K.; Selvaraj, B.T.; Barton, S.K.; Chandran, S.; Smith, C. Dysregulation of AMPA Receptor Subunit Expression in Sporadic ALS Post-mortem Brain. J. Pathol. 2020, 250, 67–78. [Google Scholar] [CrossRef]
- Rifai, O.M.; Longden, J.; O’Shaughnessy, J.; Sewell, M.D.E.; Pate, J.; McDade, K.; Daniels, M.J.D.; Abrahams, S.; Chandran, S.; McColl, B.W. Random Forest Modelling Demonstrates Microglial and Protein Misfolding Features to Be Key Phenotypic Markers in C9orf72-ALS. J. Pathol. 2022, 258, 366–381. [Google Scholar] [CrossRef]
- Weinreich, M.; Shepheard, S.R.; Verber, N.; Wyles, M.; Heath, P.R.; Highley, J.R.; Kirby, J.; Shaw, P.J. Neuropathological Characterization of a Novel TANK Binding Kinase (TBK1) Gene Loss of Function Mutation Associated with Amyotrophic Lateral Sclerosis. Neuropathol. Appl. Neurobiol. 2020, 46, 279–291. [Google Scholar] [CrossRef]
- Guerrero, E.N.; Wang, H.; Mitra, J.; Hegde, P.M.; Stowell, S.E.; Liachko, N.F.; Kraemer, B.C.; Garruto, R.M.; Rao, K.S.; Hegde, M.L. TDP-43/FUS in Motor Neuron Disease: Complexity and Challenges. Prog. Neurobiol. 2016, 145, 78–97. [Google Scholar] [CrossRef]
- Källstig, E.; McCabe, B.D.; Schneider, B.L. The Links between ALS and NF-ΚB. Int. J. Mol. Sci. 2021, 22, 3875. [Google Scholar] [CrossRef] [PubMed]
- Dąbek, J.; Kułach, A.; Gąsior, Z. Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-ΚB): A New Potential Therapeutic Target in Atherosclerosis? Pharmacol. Rep. 2010, 62, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes Expressing ALS-linked Mutant FUS Induce Motor Neuron Death through Release of Tumor Necrosis Factor-alpha. Glia 2018, 66, 1016–1033. [Google Scholar] [CrossRef] [PubMed]
- Molnar-Kasza, A.; Hinteregger, B.; Neddens, J.; Rabl, R.; Flunkert, S.; Hutter-Paier, B. Evaluation of Neuropathological Features in the SOD1-G93A Low Copy Number Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2021, 14, 681868. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P. Microglia Induce Motor Neuron Death via the Classical NF-ΚB Pathway in Amyotrophic Lateral Sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Lino, M.M.; Schneider, C.; Caroni, P. Accumulation of SOD1 Mutants in Postnatal Motoneurons Does Not Cause Motoneuron Pathology or Motoneuron Disease. J. Neurosci. 2002, 22, 4825–4832. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant Superoxide Dismutase 1-Induced IL-1β Accelerates ALS Pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillee, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J. Wild-Type Nonneuronal Cells Extend Survival of SOD1 Mutant Motor Neurons in ALS Mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-Derived TGF-Β1 Accelerates Disease Progression in ALS Mice by Interfering with the Neuroprotective Functions of Microglia and T Cells. Cell Rep. 2015, 11, 592–604. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes Mediate Amyotrophic Lateral Sclerosis Progression and Survival. EMBO Mol. Med. 2013, 5, 64–79. [Google Scholar] [CrossRef]
- Chiu, I.M.; Chen, A.; Zheng, Y.; Kosaras, B.; Tsiftsoglou, S.A.; Vartanian, T.K.; Brown, R.H., Jr.; Carroll, M.C. T Lymphocytes Potentiate Endogenous Neuroprotective Inflammation in a Mouse Model of ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 17913–17918. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D. Mouse Models of Frontotemporal Dementia. Ann. Neurol. 2012, 72, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Banerjee, R.; Thomas, B.; Zhou, P.; Qian, L.; Jia, T.; Ma, X.; Ma, Y.; Iadecola, C.; Beal, M.F. Exaggerated Inflammation, Impaired Host Defense, and Neuropathology in Progranulin-Deficient Mice. J. Exp. Med. 2010, 207, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 Mutant Transgenic Mice Develop Features of ALS and Frontotemporal Lobar Degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Ridler, C. Loss of TDP-43 in Microglia—Friend or Foe? Nat. Rev. Neurol. 2017, 13, 449. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Jawaid, A.; Henstridge, C.M.; Valeri, A.; Merlini, M.; Robinson, J.L.; Lee, E.B.; Rose, J.; Appel, S.; Lee, V.M.-Y. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron 2017, 95, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Lall, D.; Lorenzini, I.; Mota, T.A.; Bell, S.; Mahan, T.E.; Ulrich, J.D.; Davtyan, H.; Rexach, J.E.; Muhammad, A.K.M.G.; Shelest, O. C9orf72 Deficiency Promotes Microglial-Mediated Synaptic Loss in Aging and Amyloid Accumulation. Neuron 2021, 109, 2275–2291. [Google Scholar] [CrossRef]
- Swift, I.J.; Sogorb-Esteve, A.; Heller, C.; Synofzik, M.; Otto, M.; Graff, C.; Galimberti, D.; Todd, E.; Heslegrave, A.J.; van der Ende, E.L. Fluid Biomarkers in Frontotemporal Dementia: Past, Present and Future. J. Neurol. Neurosurg. Psychiatry 2021, 92, 204–215. [Google Scholar] [CrossRef]
- Hansson, O.; Lehmann, S.; Otto, M.; Zetterberg, H.; Lewczuk, P. Advantages and Disadvantages of the Use of the CSF Amyloid β (Aβ) 42/40 Ratio in the Diagnosis of Alzheimer’s Disease. Alzheimers Res. Ther. 2019, 11, 1–15. [Google Scholar] [CrossRef]
- Pouclet-Courtemanche, H.; Nguyen, T.-B.; Skrobala, E.; Boutoleau-Bretonnière, C.; Pasquier, F.; Bouaziz-Amar, E.; Bigot-Corbel, E.; Schraen, S.; Dumurgier, J.; Paquet, C. Frontotemporal Dementia Is the Leading Cause of “True” A−/T+ Profiles Defined with Aβ42/40 Ratio. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 161–169. [Google Scholar]
- Chiu, P.-Y.; Yang, F.-C.; Chiu, M.-J.; Lin, W.-C.; Lu, C.-H.; Yang, S.-Y. Relevance of Plasma Biomarkers to Pathologies in Alzheimer’s Disease, Parkinson’s Disease and Frontotemporal Dementia. Sci. Rep. 2022, 12, 17919. [Google Scholar] [CrossRef] [PubMed]
- Katzeff, J.S.; Bright, F.; Phan, K.; Kril, J.J.; Ittner, L.M.; Kassiou, M.; Hodges, J.R.; Piguet, O.; Kiernan, M.C.; Halliday, G.M. Biomarker Discovery and Development for Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Brain 2022, 145, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament Light Chain as a Biomarker in Neurological Disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Bridel, C.; Van Wieringen, W.N.; Zetterberg, H.; Tijms, B.M.; Teunissen, C.E.; Alvarez-Cermeño, J.C.; Andreasson, U.; Axelsson, M.; Bäckström, D.C.; Bartos, A. Diagnostic Value of Cerebrospinal Fluid Neurofilament Light Protein in Neurology: A Systematic Review and Meta-Analysis. JAMA Neurol. 2019, 76, 1035–1048. [Google Scholar] [CrossRef] [PubMed]
- Verde, F.; Steinacker, P.; Weishaupt, J.H.; Kassubek, J.; Oeckl, P.; Halbgebauer, S.; Tumani, H.; von Arnim, C.A.F.; Dorst, J.; Feneberg, E. Neurofilament Light Chain in Serum for the Diagnosis of Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Qin, X.; Chang, X.; Wang, H.; Guo, J.; Zhang, W. Neurofilament Markers in Serum and Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis. J. Cell Mol. Med. 2022, 26, 583–587. [Google Scholar] [CrossRef]
- Landqvist Waldö, M.; Frizell Santillo, A.; Passant, U.; Zetterberg, H.; Rosengren, L.; Nilsson, C.; Englund, E. Cerebrospinal Fluid Neurofilament Light Chain Protein Levels in Subtypes of Frontotemporal Dementia. BMC Neurol. 2013, 13, 1–8. [Google Scholar] [CrossRef]
- Ooi, S.; Patel, S.K.; Eratne, D.; Kyndt, C.; Reidy, N.; Lewis, C.; Lee, S.; Darby, D.; Brodtmann, A. Plasma Neurofilament Light Chain and Clinical Diagnosis in Frontotemporal Dementia Syndromes. J. Alzheimer’s Dis. 2022, 1–11, preprint. [Google Scholar] [CrossRef]
- Spotorno, N.; Lindberg, O.; Nilsson, C.; Landqvist Waldö, M.; Van Westen, D.; Nilsson, K.; Vestberg, S.; Englund, E.; Zetterberg, H.; Blennow, K. Plasma Neurofilament Light Protein Correlates with Diffusion Tensor Imaging Metrics in Frontotemporal Dementia. PLoS ONE 2020, 15, e0236384. [Google Scholar] [CrossRef]
- Saracino, D.; Dorgham, K.; Camuzat, A.; Rinaldi, D.; Rametti-Lacroux, A.; Houot, M.; Clot, F.; Martin-Hardy, P.; Jornea, L.; Azuar, C. Plasma NfL Levels and Longitudinal Change Rates in C9orf72 and GRN-Associated Diseases: From Tailored References to Clinical Applications. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Meeter, L.H.; Dopper, E.G.; Jiskoot, L.C.; Sanchez-Valle, R.; Graff, C.; Benussi, L.; Ghidoni, R.; Pijnenburg, Y.A.; Borroni, B.; Galimberti, D. Neurofilament Light Chain: A Biomarker for Genetic Frontotemporal Dementia. Ann. Clin. Transl. Neurol. 2016, 3, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhu, Y.; Hsiao-Nakamoto, J.; Tang, X.; Dugas, J.C.; Moscovitch-Lopatin, M.; Glass, J.D.; Brown, R.H., Jr.; Ladha, S.S.; Lacomis, D. Longitudinal Biomarkers in Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Béland, L.-C.; Markovinovic, A.; Jakovac, H.; de Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in Amyotrophic Lateral Sclerosis: Blurred Lines between Excessive Inflammation and Inefficient Immune Responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Munitic, I.; Amedei, A.; Berry, J.D.; Feldman, E.L.; Aronica, E.; Nardo, G.; Van Weehaeghe, D.; Niccolai, E.; Prtenjaca, N. Interplay between Immunity and Amyotrophic Lateral Sclerosis: Clinical Impact. Neurosci. Biobehav. Rev. 2021, 127, 958–978. [Google Scholar] [CrossRef]
- Renkema, G.H.; Boot, R.G.; Au, F.L.; Donker-Koopman, W.E.; Strijland, A.; Muijsers, A.O.; Hrebicek, M.; Aerts, J.M.F.G. Chitotriosidase, a Chitinase, and the 39-kDa Human Cartilage Glycoprotein, a Chitin-binding Lectin, Are Homologues of Family 18 Glycosyl Hydrolases Secreted by Human Macrophages. Eur. J. Biochem. 1998, 251, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, M.; Egashira, K.; Kitamoto, S.; Ni, W.; Shimokawa, H.; Takeya, M.; Yoshimura, T.; Takeshita, A. Role of Monocyte Chemoattractant Protein-1 in Cardiovascular Remodeling Induced by Chronic Blockade of Nitric Oxide Synthesis. Circulation 2000, 102, 2243–2248. [Google Scholar] [CrossRef]
- Oeckl, P.; Weydt, P.; Steinacker, P.; Anderl-Straub, S.; Nordin, F.; Volk, A.E.; Diehl-Schmid, J.; Andersen, P.M.; Kornhuber, J.; Danek, A. Different Neuroinflammatory Profile in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Is Linked to the Clinical Phase. J. Neurol. Neurosurg. Psychiatry 2019, 90, 4–10. [Google Scholar] [CrossRef]
- Barschke, P.; Oeckl, P.; Steinacker, P.; Al Shweiki, M.H.D.R.; Weishaupt, J.H.; Landwehrmeyer, G.B.; Anderl-Straub, S.; Weydt, P.; Diehl-Schmid, J.; Danek, A. Different CSF Protein Profiles in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia with C9orf72 Hexanucleotide Repeat Expansion. J. Neurol. Neurosurg. Psychiatry 2020, 91, 503–511. [Google Scholar] [CrossRef]
- Heller, C.; Foiani, M.S.; Moore, K.; Convery, R.; Bocchetta, M.; Neason, M.; Cash, D.M.; Thomas, D.; Greaves, C.V.; Woollacott, I.O.; et al. Plasma Glial Fibrillary Acidic Protein Is Raised in Progranulin-Associated Frontotemporal Dementia. J. Neurol. Neurosurg. Psychiatry 2020, 91, 263–270. [Google Scholar] [CrossRef]
- Galimberti, D.; Bonsi, R.; Fenoglio, C.; Serpente, M.; Cioffi, S.M.G.; Fumagalli, G.; Arighi, A.; Ghezzi, L.; Arcaro, M.; Mercurio, M.; et al. Inflammatory Molecules in Frontotemporal Dementia: Cerebrospinal Fluid Signature of Progranulin Mutation Carriers. Brain Behav. Immun. 2015, 49, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Mazaheri, F.; Snaidero, N.; Kleinberger, G.; Madore, C.; Daria, A.; Werner, G.; Krasemann, S.; Capell, A.; Trümbach, D.; Wurst, W. TREM 2 Deficiency Impairs Chemotaxis and Microglial Responses to Neuronal Injury. EMBO Rep. 2017, 18, 1186–1198. [Google Scholar] [CrossRef] [PubMed]
- Öhrfelt, A.; Axelsson, M.; Malmeström, C.; Novakova, L.; Heslegrave, A.; Blennow, K.; Lycke, J.; Zetterberg, H. Soluble TREM-2 in Cerebrospinal Fluid from Patients with Multiple Sclerosis Treated with Natalizumab or Mitoxantrone. Mult. Scler. J. 2016, 22, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Carmona, S.; Zahs, K.; Wu, E.; Dakin, K.; Bras, J.; Guerreiro, R. The Role of TREM2 in Alzheimer’s Disease and Other Neurodegenerative Disorders. Lancet Neurol. 2018, 17, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Morenas-Rodríguez, E.; Li, Y.; Nuscher, B.; Franzmeier, N.; Xiong, C.; Suárez-Calvet, M.; Fagan, A.M.; Schultz, S.; Gordon, B.A.; Benzinger, T.L.S. Soluble TREM2 in CSF and Its Association with Other Biomarkers and Cognition in Autosomal-Dominant Alzheimer’s Disease: A Longitudinal Observational Study. Lancet Neurol. 2022, 21, 329–341. [Google Scholar] [CrossRef]
- Paloneva, J.; Kestilä, M.; Wu, J.; Salminen, A.; Böhling, T.; Ruotsalainen, V.; Hakola, P.; Bakker, A.B.H.; Phillips, J.H.; Pekkarinen, P. Loss-of-Function Mutations in TYROBP (DAP12) Result in a Presenile Dementia with Bone Cysts. Nat. Genet. 2000, 25, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Le Ber, I.; De Septenville, A.; Guerreiro, R.; Bras, J.; Camuzat, A.; Caroppo, P.; Lattante, S.; Couarch, P.; Kabashi, E.; Bouya-Ahmed, K. Homozygous TREM2 Mutation in a Family with Atypical Frontotemporal Dementia. Neurobiol. Aging 2014, 35, 2419.e23–2419.e25. [Google Scholar] [CrossRef]
- Woollacott, I.O.C.; Nicholas, J.M.; Heslegrave, A.; Heller, C.; Foiani, M.S.; Dick, K.M.; Russell, L.L.; Paterson, R.W.; Keshavan, A.; Fox, N.C. Cerebrospinal Fluid Soluble TREM2 Levels in Frontotemporal Dementia Differ by Genetic and Pathological Subgroup. Alzheimers Res. Ther. 2018, 10, 1–14. [Google Scholar] [CrossRef]
- Ghidoni, R.; Stoppani, E.; Rossi, G.; Piccoli, E.; Albertini, V.; Paterlini, A.; Glionna, M.; Pegoiani, E.; Agnati, L.F.; Fenoglio, C. Optimal Plasma Progranulin Cutoff Value for Predicting Null Progranulin Mutations in Neurodegenerative Diseases: A Multicenter Italian Study. Neurodegener. Dis. 2012, 9, 121–127. [Google Scholar] [CrossRef]
- Sellami, L.; Rucheton, B.; Ben Younes, I.; Camuzat, A.; Saracino, D.; Rinaldi, D.; Epelbaum, S.; Azuar, C.; Levy, R.; Auriacombe, S. Plasma Progranulin Levels for Frontotemporal Dementia in Clinical Practice: A 10-Year French Experience. Neurobiol. Aging 2020, 91, 167.e1–167.e9. [Google Scholar] [CrossRef]
- Steinacker, P.; Hendrich, C.; Sperfeld, A.D.; Jesse, S.; von Arnim, C.A.F.; Lehnert, S.; Pabst, A.; Uttner, I.; Tumani, H.; Lee, V.M.-Y.; et al. TDP-43 in Cerebrospinal Fluid of Patients with Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Arch. Neurol. 2008, 65, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Foulds, P.; McAuley, E.; Gibbons, L.; Davidson, Y.; Pickering-Brown, S.M.; Neary, D.; Snowden, J.S.; Allsop, D.; Mann, D.M.A. TDP-43 Protein in Plasma May Index TDP-43 Brain Pathology in Alzheimer’s Disease and Frontotemporal Lobar Degeneration. Acta Neuropathol. 2008, 116, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of Primary Progressive Aphasia and Its Variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.M.; Bejanin, A.; Lesman-Segev, O.; LaJoie, R.; Visani, A.; Bourakova, V.; O’Neil, J.P.; Janabi, M.; Baker, S.; Lee, S.E. 18 F-Flortaucipir (AV-1451) Tau PET in Frontotemporal Dementia Syndromes. Alzheimers Res. Ther. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-H.; Chen, T.-F.; Chiu, M.-J.; Yen, R.-F.; Shih, M.-C.; Lin, C.-H. Integrated 18F-T807 Tau PET, Structural MRI, and Plasma Tau in Tauopathy Neurodegenerative Disorders. Front. Aging Neurosci. 2021, 13, 646440. [Google Scholar] [CrossRef] [PubMed]
- Agosta, F.; Altomare, D.; Festari, C.; Orini, S.; Gandolfo, F.; Boccardi, M.; Arbizu, J.; Bouwman, F.; Drzezga, A.; Nestor, P. Clinical Utility of FDG-PET in Amyotrophic Lateral Sclerosis and Huntington’s Disease. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, V.; Pioro, E.P. Comparing Brain Structural MRI and Metabolic FDG-PET Changes in Patients with ALS-FTD:‘The Chicken or the Egg?’Question. J. Neurol. Neurosurg. Psychiatry 2015, 86, 952–958. [Google Scholar] [CrossRef]
- De Marchi, F.; Stecco, A.; Falaschi, Z.; Filippone, F.; Pasché, A.; Bebeti, A.; Leigheb, M.; Cantello, R.; Mazzini, L. Detection of White Matter Ultrastructural Changes for Amyotrophic Lateral Sclerosis Characterization: A Diagnostic Study from Dti-Derived Data. Brain Sci. 2020, 10, 996. [Google Scholar] [CrossRef]
- Canosa, A.; Pagani, M.; Cistaro, A.; Montuschi, A.; Iazzolino, B.; Fania, P.; Cammarosano, S.; Ilardi, A.; Moglia, C.; Calvo, A.; et al. 18 F-FDG-PET Correlates of Cognitive Impairment in ALS. Neurology 2016, 86, 44–49. [Google Scholar] [CrossRef]
- Dadar, M.; Manera, A.L.; Zinman, L.; Korngut, L.; Genge, A.; Graham, S.J.; Frayne, R.; Collins, D.L.; Kalra, S. Cerebral Atrophy in Amyotrophic Lateral Sclerosis Parallels the Pathological Distribution of TDP43. Brain Commun. 2020, 2, fcaa061. [Google Scholar] [CrossRef]
- Tondo, G.; Mazzini, L.; Caminiti, S.P.; Sarnelli, M.F.; Corrado, L.; Matheoud, R.; D’Alfonso, S.; Cantello, R.; Sacchetti, G.M.; Perani, D. Clinical Relevance of Single-Subject Brain Metabolism Patterns in Amyotrophic Lateral Sclerosis Mutation Carriers. Neuroimage Clin. 2022, 36, 103222. [Google Scholar] [CrossRef] [PubMed]
- De Vocht, J.; Van Weehaeghe, D.; Ombelet, F.; Masrori, P.; Lamaire, N.; Devrome, M.; Van Esch, H.; Moisse, M.; Koole, M.; Dupont, P. Differences in Cerebral Glucose Metabolism in ALS Patients with and without C9orf72 and SOD1 Mutations. Cells 2023, 12, 933. [Google Scholar] [CrossRef]
- Spinelli, E.G.; Ghirelli, A.; Basaia, S.; Cividini, C.; Riva, N.; Canu, E.; Castelnovo, V.; Domi, T.; Magnani, G.; Caso, F. Structural MRI Signatures in Genetic Presentations of the Frontotemporal Dementia/Motor Neuron Disease Spectrum. Neurology 2021, 97, e1594–e1607. [Google Scholar] [CrossRef] [PubMed]
- Tondo, G.; Boccalini, C.; Caminiti, S.P.; Presotto, L.; Filippi, M.; Magnani, G.; Frisoni, G.B.; Iannaccone, S.; Perani, D. Brain Metabolism and Microglia Activation in Mild Cognitive Impairment: A Combined [18F] FDG and [11C]-(R)-PK11195 PET Study. J. Alzheimer’s Dis. 2021, 80, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, L.; Rodríguez, P.V.; Hong, Y.T.; Allinson, K.S.J.; Bevan-Jones, W.R.; Williamson, D.; Jones, P.S.; Arnold, R.; Borchert, R.J.; Surendranathan, A. [11C] PK11195 Binding in Alzheimer Disease and Progressive Supranuclear Palsy. Neurology 2018, 90, e1989–e1996. [Google Scholar] [CrossRef] [PubMed]
- Kübler, D.; Wächter, T.; Cabanel, N.; Su, Z.; Turkheimer, F.E.; Dodel, R.; Brooks, D.J.; Oertel, W.H.; Gerhard, A. Widespread Microglial Activation in Multiple System Atrophy. Mov. Disord. 2019, 34, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Schain, M.; Kreisl, W.C. Neuroinflammation in Neurodegenerative Disorders—A Review. Curr. Neurol. Neurosci. Rep. 2017, 17, 1–11. [Google Scholar] [CrossRef]
- Cagnin, A.; Rossor, M.; Sampson, E.L.; MacKinnon, T.; Banati, R.B. In Vivo Detection of Microglial Activation in Frontotemporal Dementia. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 2004, 56, 894–897. [Google Scholar] [CrossRef]
- Zhang, J. Mapping Neuroinflammation in Frontotemporal Dementia with Molecular PET Imaging. J. Neuroinflammation 2015, 12, 1–7. [Google Scholar] [CrossRef]
- Miyoshi, M.; Shinotoh, H.; Wszolek, Z.K.; Strongosky, A.J.; Shimada, H.; Arakawa, R.; Higuchi, M.; Ikoma, Y.; Yasuno, F.; Fukushi, K. In Vivo Detection of Neuropathologic Changes in Presymptomatic MAPT Mutation Carriers: A PET and MRI Study. Park. Relat. Disord. 2010, 16, 404–408. [Google Scholar] [CrossRef]
- Lant, S.B.; Robinson, A.C.; Thompson, J.C.; Rollinson, S.; Pickering-Brown, S.; Snowden, J.S.; Davidson, Y.S.; Gerhard, A.; Mann, D.M.A. Patterns of Microglial Cell Activation in Frontotemporal Lobar Degeneration. Neuropathol. Appl. Neurobiol. 2014, 40, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Malpetti, M.; Rittman, T.; Jones, P.S.; Cope, T.E.; Passamonti, L.; Bevan-Jones, W.R.; Patterson, K.; Fryer, T.D.; Hong, Y.T.; Aigbirhio, F.I. In Vivo PET Imaging of Neuroinflammation in Familial Frontotemporal Dementia. J. Neurol. Neurosurg. Psychiatry 2021, 92, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Malpetti, M.; Cope, T.E.; Street, D.; Jones, P.S.; Hezemans, F.H.; Mak, E.; Tsvetanov, K.A.; Rittman, T.; Bevan-Jones, W.R.; Patterson, K. Microglial Activation in the Frontal Cortex Predicts Cognitive Decline in Frontotemporal Dementia. Brain 2023, 146, 3221–3231. [Google Scholar] [CrossRef]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of Widespread Cerebral Microglial Activation in Amyotrophic Lateral Sclerosis: An [11C](R)-PK11195 Positron Emission Tomography Study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Alshikho, M.J.; Zürcher, N.R.; Loggia, M.L.; Cernasov, P.; Reynolds, B.; Pijanowski, O.; Chonde, D.B.; Izquierdo Garcia, D.; Mainero, C.; Catana, C. Integrated Magnetic Resonance Imaging and [11C]-PBR28 Positron Emission Tomographic Imaging in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2018, 83, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Tauber, C.; Vercoullie, J.; Arlicot, N.; Prunier, C.; Praline, J.; Nicolas, G.; Venel, Y.; Hommet, C.; Baulieu, J.-L. Molecular Imaging of Microglial Activation in Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e52941. [Google Scholar] [CrossRef] [PubMed]
- Alshikho, M.J.; Zürcher, N.R.; Loggia, M.L.; Cernasov, P.; Chonde, D.B.; Garcia, D.I.; Yasek, J.E.; Akeju, O.; Catana, C.; Rosen, B.R. Glial Activation Colocalizes with Structural Abnormalities in Amyotrophic Lateral Sclerosis. Neurology 2016, 87, 2554–2561. [Google Scholar] [CrossRef]
- Zürcher, N.R.; Loggia, M.L.; Lawson, R.; Chonde, D.B.; Izquierdo-Garcia, D.; Yasek, J.E.; Akeju, O.; Catana, C.; Rosen, B.R.; Cudkowicz, M.E. Increased in Vivo Glial Activation in Patients with Amyotrophic Lateral Sclerosis: Assessed with [11C]-PBR28. Neuroimage Clin. 2015, 7, 409–414. [Google Scholar] [CrossRef]
- Ratai, E.-M.; Alshikho, M.J.; Zürcher, N.R.; Loggia, M.L.; Cebulla, C.L.; Cernasov, P.; Reynolds, B.; Fish, J.; Seth, R.; Babu, S. Integrated Imaging of [11C]-PBR28 PET, MR Diffusion and Magnetic Resonance Spectroscopy 1H-MRS in Amyotrophic Lateral Sclerosis. Neuroimage Clin. 2018, 20, 357–364. [Google Scholar] [CrossRef]
- Tondo, G.; Iaccarino, L.; Cerami, C.; Vanoli, G.E.; Presotto, L.; Masiello, V.; Coliva, A.; Salvi, F.; Bartolomei, I.; Mosca, L. 11C-PK11195 PET–Based Molecular Study of Microglia Activation in SOD1 Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1513–1523. [Google Scholar] [CrossRef]
- Turner, M.R.; Goldacre, R.; Ramagopalan, S.; Talbot, K.; Goldacre, M.J. Autoimmune Disease Preceding Amyotrophic Lateral Sclerosis: An Epidemiologic Study. Neurology 2013, 81, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Huang, W.; Cao, F.; Yu, X.; Guo, S.; Ying, Z.; Xu, C. Causal Association between Systemic Lupus Erythematosus and the Risk of Dementia: A Mendelian Randomization Study. Front. Immunol. 2022, 13, 1063110. [Google Scholar] [CrossRef] [PubMed]
- Miller, Z.A.; Rankin, K.P.; Graff-Radford, N.R.; Takada, L.T.; Sturm, V.E.; Cleveland, C.M.; Criswell, L.A.; Jaeger, P.A.; Stan, T.; Heggeli, K.A. TDP-43 Frontotemporal Lobar Degeneration and Autoimmune Disease. J. Neurol. Neurosurg. Psychiatry 2013, 84, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Miller, Z.A.; Sturm, V.E.; Camsari, G.B.; Karydas, A.; Yokoyama, J.S.; Grinberg, L.T.; Boxer, A.L.; Rosen, H.J.; Rankin, K.P.; Gorno-Tempini, M.L. Increased Prevalence of Autoimmune Disease within C9 and FTD/MND Cohorts: Completing the Picture. Neurol. -Neuroimmunol. Neuroinflammation 2016, 3, e301. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Cooper-Knock, J.; Highley, J.R.; Milano, A.; Kirby, J.; Goodall, E.; Lowe, J.; Scott, I.; Constantinescu, C.S.; Walters, S.J. Concurrence of Multiple Sclerosis and Amyotrophic Lateral Sclerosis in Patients with Hexanucleotide Repeat Expansions of C9ORF72. J. Neurol. Neurosurg. Psychiatry 2013, 84, 79–87. [Google Scholar] [CrossRef]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491. [Google Scholar] [CrossRef]
- Choi, S.-J.; Hong, Y.-H.; Kim, S.-M.; Shin, J.-Y.; Suh, Y.J.; Sung, J.-J. High Neutrophil-to-Lymphocyte Ratio Predicts Short Survival Duration in Amyotrophic Lateral Sclerosis. Sci. Rep. 2020, 10, 428. [Google Scholar] [CrossRef]
- Wei, Q.-Q.; Hou, Y.-B.; Zhang, L.-Y.; Ou, R.-W.; Cao, B.; Chen, Y.-P.; Shang, H.-F. Neutrophil-to-Lymphocyte Ratio in Sporadic Amyotrophic Lateral Sclerosis. Neural Regen. Res. 2022, 17, 875. [Google Scholar]
- Leone, M.A.; Mandrioli, J.; Russo, S.; Cucovici, A.; Gianferrari, G.; Lisnic, V.; Muresanu, D.F.; Giuliani, F.; Copetti, M.; Consortium, P.R.O.-A.A.L.S.C.T. Neutrophils-to-Lymphocyte Ratio Is Associated with Progression and Overall Survival in Amyotrophic Lateral Sclerosis. Biomedicines 2022, 10, 354. [Google Scholar] [CrossRef]
- Lunetta, C.; Lizio, A.; Maestri, E.; Sansone, V.A.; Mora, G.; Miller, R.G.; Appel, S.H.; Chiò, A. Serum C-Reactive Protein as a Prognostic Biomarker in Amyotrophic Lateral Sclerosis. JAMA Neurol. 2017, 74, 660–667. [Google Scholar] [CrossRef]
- Beers, D.R.; Zhao, W.; Neal, D.W.; Thonhoff, J.R.; Thome, A.D.; Faridar, A.; Wen, S.; Wang, J.; Appel, S.H. Elevated Acute Phase Proteins Reflect Peripheral Inflammation and Disease Severity in Patients with Amyotrophic Lateral Sclerosis. Sci. Rep. 2020, 10, 15295. [Google Scholar] [CrossRef]
- Tanaka, H.; Shimazawa, M.; Kimura, M.; Takata, M.; Tsuruma, K.; Yamada, M.; Takahashi, H.; Hozumi, I.; Niwa, J.; Iguchi, Y. The Potential of GPNMB as Novel Neuroprotective Factor in Amyotrophic Lateral Sclerosis. Sci. Rep. 2012, 2, 573. [Google Scholar] [CrossRef]
- Conte, A.; Lattante, S.; Zollino, M.; Marangi, G.; Luigetti, M.; Del Grande, A.; Servidei, S.; Trombetta, F.; Sabatelli, M. P525L FUS Mutation Is Consistently Associated with a Severe Form of Juvenile Amyotrophic Lateral Sclerosis. Neuromuscul. Disord. 2012, 22, 73–75. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Marchi, F.; Tondo, G.; Corrado, L.; Menegon, F.; Aprile, D.; Anselmi, M.; D’Alfonso, S.; Comi, C.; Mazzini, L. Neuroinflammatory Pathways in the ALS-FTD Continuum: A Focus on Genetic Variants. Genes 2023, 14, 1658. https://doi.org/10.3390/genes14081658
De Marchi F, Tondo G, Corrado L, Menegon F, Aprile D, Anselmi M, D’Alfonso S, Comi C, Mazzini L. Neuroinflammatory Pathways in the ALS-FTD Continuum: A Focus on Genetic Variants. Genes. 2023; 14(8):1658. https://doi.org/10.3390/genes14081658
Chicago/Turabian StyleDe Marchi, Fabiola, Giacomo Tondo, Lucia Corrado, Federico Menegon, Davide Aprile, Matteo Anselmi, Sandra D’Alfonso, Cristoforo Comi, and Letizia Mazzini. 2023. "Neuroinflammatory Pathways in the ALS-FTD Continuum: A Focus on Genetic Variants" Genes 14, no. 8: 1658. https://doi.org/10.3390/genes14081658
APA StyleDe Marchi, F., Tondo, G., Corrado, L., Menegon, F., Aprile, D., Anselmi, M., D’Alfonso, S., Comi, C., & Mazzini, L. (2023). Neuroinflammatory Pathways in the ALS-FTD Continuum: A Focus on Genetic Variants. Genes, 14(8), 1658. https://doi.org/10.3390/genes14081658