
The Huntington’s Disease Gene in an Italian Cohort of Patients with Bipolar Disorder

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Population
2.2. Genetic Analysis
2.3. Statistical Analysis
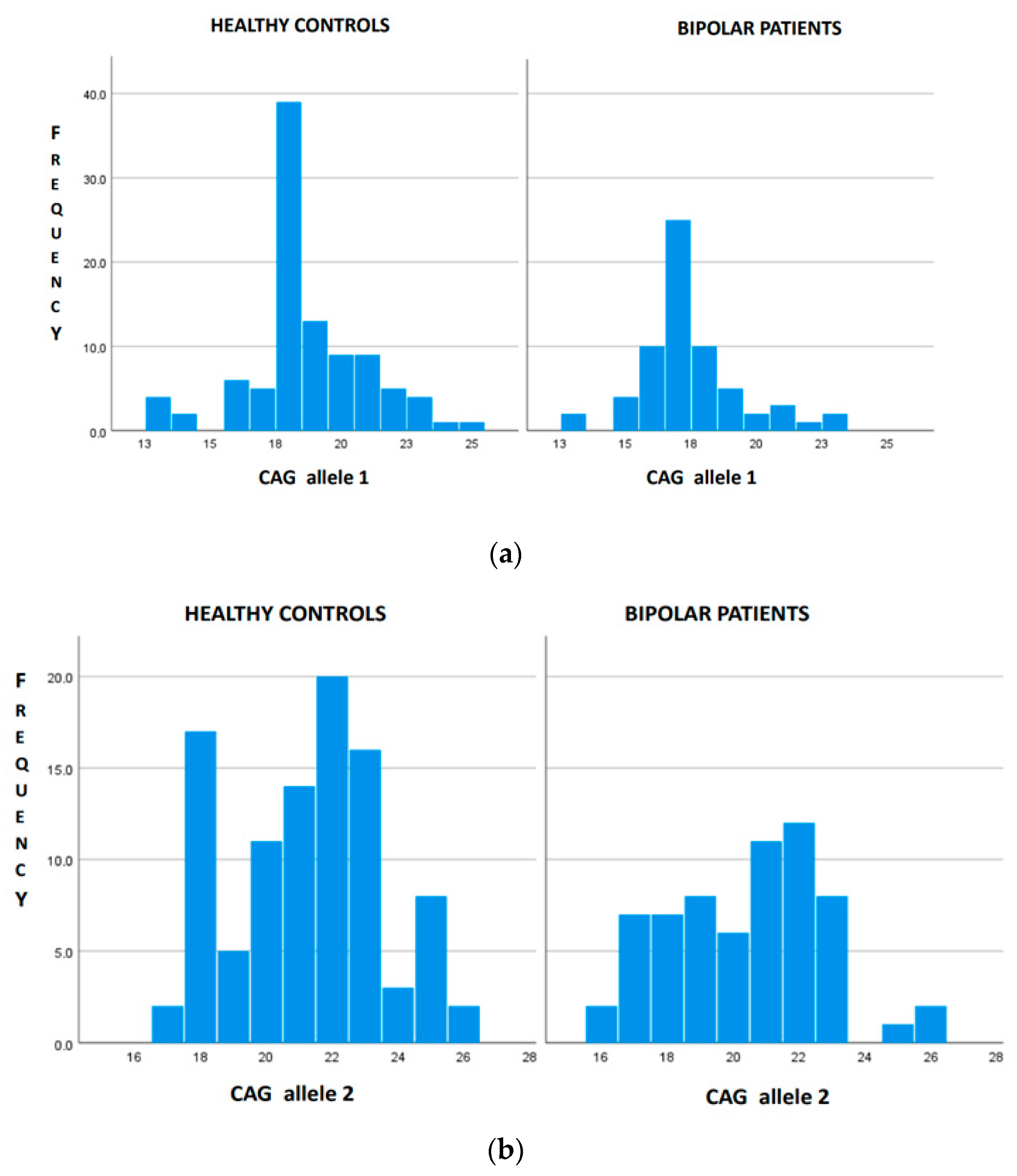
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Kay, C.; Hayden, M.R.; Leavitt, B.R. Epidemiology of Huntington disease. Handb. Clin. Neurol. 2017, 144, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Reilmann, R.; Cardoso, F.; McCusker, E.A.; Testa, C.M.; Stout, J.C.; Leavitt, B.R.; Pei, Z.; Landwehrmeyer, B.; Martinez, A.; et al. Movement Disorder Society Task Force Viewpoint: Huntington’s Disease Diagnostic Categories. Mov. Disord. Clin. Pract. 2019, 6, 541–546. [Google Scholar] [CrossRef]
- Julien, C.L.; Thompson, J.C.; Wild, S.; Yardumian, P.; Snowden, J.S.; Turner, G.; Craufurd, D. Psychiatric disorders in preclinical Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Handley, O.J.; Schwenke, C.; Dunnett, S.B.; Craufurd, D.; Ho, A.K.; Wild, E.; Tabrizi, S.J.; Landwehrmeyer, G.B. The investigators of the European Huntington’s Disease Network Observing Huntington’s Disease: The European Huntington’s Disease Network’s REGISTRY. PLoS Curr. 2010, 2, RRN1184. [Google Scholar] [CrossRef]
- Hammer, M.B.; Singleton, A.B. Common Premutations in the General Population. JAMA Neurol. 2019, 76, 639–640. [Google Scholar] [CrossRef] [PubMed]
- Apolinário, T.; Paiva, C.; Agostinho, L. REVIEW-ARTICLE Intermediate alleles of Huntington’s disease HTT gene in different populations worldwide: A systematic review. Genet. Mol. Res. 2017, 16, gmr16029648. [Google Scholar] [CrossRef]
- Sundblom, J.; Niemelä, V.; Ghazarian, M.; Strand, A.S.; Bergdahl, I.A.; Jansson, J.H.; Söderberg, S.; Stattin, E.L. High frequency of intermediary alleles in the HTT gene in Northern Sweden—The Swedish Huntingtin Alleles and Phenotype (SHAPE) study. Sci. Rep. 2020, 10, 9853. [Google Scholar] [CrossRef]
- Oosterloo, M.; Van Belzen, M.J.; Bijlsma, E.K.; Roos, R.A. Is There Convincing Evidence that Intermediate Repeats in the HTT Gene Cause Huntington’s Disease? J. Huntingt. Dis. 2015, 4, 141–148. [Google Scholar] [CrossRef]
- Killoran, A.; Biglan, K.M.; Jankovic, J.; Eberly, S.; Kayson, E.; Oakes, D.; Young, A.B.; Shoulson, I. Characterization of the Huntington intermediate CAG repeat expansion phenotype in PHAROS. Neurology 2013, 80, 2022–2027. [Google Scholar] [CrossRef]
- Kenney, C.; Powell, S.; Jankovic, J. Autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov. Disord. 2007, 22, 127–130. [Google Scholar] [CrossRef]
- Savitt, D.; Jankovic, J. Clinical phenotype in carriers of intermediate alleles in the huntingtin gene. J. Neurol. Sci. 2019, 402, 57–61. [Google Scholar] [CrossRef]
- Jevtic, S.D.; Provias, J.P. Case report and literature review of Huntington disease with intermediate CAG expansion. BMJ Neurol. Open 2020, 2, e000027. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.S.; Coombes, B.J. Genetic contributions to bipolar disorder: Current status and future directions. Psychol. Med. 2021, 51, 2156–2167. [Google Scholar] [CrossRef] [PubMed]
- Gordovez, F.J.A.; McMahon, F.J. The genetics of bipolar disorder. Mol. Psychiatry 2020, 25, 544–559. [Google Scholar] [CrossRef]
- Peyser, C.E.; Folstein, S.E. Huntington’s disease as a model for mood disorders. Clues from neuropathology and neurochemistry. Mol. Chem. Neuropathol. 1990, 12, 99–119. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F. Huntington’s disease: Update and review of neuropsychiatric aspects. Int. J. Psychiatry Med. 1994, 24, 189–208. [Google Scholar] [CrossRef]
- Blumenstock, S.; Dudanova, I. Cortical and Striatal Circuits in Huntington’s Disease. Front. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Garcia-Gorro, C.; Llera, A.; Martinez-Horta, S.; Perez-Perez, J.; Kulisevsky, J.; Rodriguez-Dechicha, N.; Vaquer, I.; Subira, S.; Calopa, M.; Muñoz, E.; et al. Specific patterns of brain alterations underlie distinct clinical profiles in Huntington’s disease. NeuroImage Clin. 2019, 23, 101900. [Google Scholar] [CrossRef]
- Phillips, M.L.; Swartz, H.A.; Sharma, A.; Wolf, D.H.; Ciric, R.; Kable, J.W.; Moore, T.M.; Vandekar, S.N.; Katchmar, N.; Daldal, A.; et al. A Critical Appraisal of Neuroimaging Studies of Bipolar Disorder: Toward a New Conceptualization of Underlying Neural Circuitry and a Road Map for Future Research. Am. J. Psychiatry 2014, 171, 829–843. [Google Scholar] [CrossRef]
- Diaz, A.P.; Bauer, I.E.; Sanches, M.; Soares, J.C. Neuroanatomic and Functional Neuroimaging Findings. Curr. Top. Behav. Neurosci. 2021, 48, 173–196. [Google Scholar] [CrossRef] [PubMed]
- Young, A.H.; Juruena, M.F. The Neurobiology of Bipolar Disorder. Curr. Top. Behav. Neurosci. 2021, 48, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular Mechanisms and Potential Therapeutical Targets in Huntington’s Disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- Gardiner, S.L.; Van Belzen, M.J.; Boogaard, M.W.; van Roon-Mom, W.M.; Rozing, M.P.; Van Hemert, A.M.; Smit, J.H.; Beekman, A.T.; Van Grootheest, G.; Schoevers, R.A.; et al. Huntingtin gene repeat size variations affect risk of lifetime depression. Transl. Psychiatry 2017, 7, 1277. [Google Scholar] [CrossRef] [PubMed]
- Perlis, R.H.; Smoller, J.W.; Mysore, J.; Sun, M.; Gillis, T.; Purcell, S.; Rietschel, M.; Nöthen, M.M.; Witt, S.; Maier, W.; et al. Prevalence of incompletely penetrant Huntington’s disease alleles among individuals with major depressive disorder. Am. J. Psychiatry 2010, 167, 574–579. [Google Scholar] [CrossRef]
- Pang, T.Y.; Du, X.; Zajac, M.S.; Howard, M.L.; Hannan, A.J. Altered serotonin receptor expression is associated with depression-related behavior in the R6/1 transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 2009, 18, 753–766. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; American Psychiatric Association: Washington, DC, USA, 2000. [Google Scholar] [CrossRef]
- Ingannato, A.; Bagnoli, S.; Bessi, V.; Ferrari, C.; Mazzeo, S.; Sorbi, S.; Nacmias, B. Intermediate alleles of HTT: A new pathway in longevity. J. Neurol. Sci. 2022, 438, 120274. [Google Scholar] [CrossRef]
- Jama, M.; Millson, A.; Miller, C.E.; Lyon, E. Triplet Repeat Primed PCR Simplifies Testing for Huntington Disease. J. Mol. Diagn. 2013, 15, 255–262. [Google Scholar] [CrossRef]
- Ramos, E.M.; Gillis, T.; Mysore, J.S.; Lee, J.M.; Alonso, I.; Gusella, J.F.; Smoller, J.W.; Sklar, P.; MacDonald, M.E.; Perlis, R.H. Prevalence of Huntington’s disease gene CAG trinucleotide repeat alleles in patients with bipolar disorder. Bipolar. Disord. 2015, 17, 403–408. [Google Scholar] [CrossRef]
- Raskin, S.; Allan, N.; Teive, H.A.; Cardoso, F.; Haddad, M.S.; Levi, G.; Boy, R.; Lerena, J.U.A.N., Jr.; Sotomaior, V.S.; Janzen-Dück, M.Ô.N.I.C.A.; et al. Huntington disease: DNA analysis in Brazilian population. Arq. Neuropsiquiatr. 2000, 58, 977–985. [Google Scholar] [CrossRef]
- Lee, J.K.; Conrad, A.; Epping, E.; Mathews, K.; Magnotta, V.; Dawson, J.D.; Nopoulos, P. Effect of Trinucleotide Repeats in the Huntington’s Gene on Intelligence. EBioMedicine 2018, 3, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Hannan, A.J. Tandem Repeat Polymorphisms: Genetic Plasticity, Neural Diversity and Disease; Landes Bioscience and Springer Scienc: New York, NY, USA, 2012. [Google Scholar]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Tartari, M.; Gissi, C.; Sardo, V.L.; Zuccato, C.; Picardi, E.; Pesole, G.; Cattaneo, E. Phylogenetic Comparison of Huntingtin Homologues Reveals the Appearance of a Primitive polyQ in Sea Urchin. Mol. Biol. Evol. 2008, 25, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Mühlau, M.; Winkelmann, J.; Rujescu, D.; Giegling, I.; Koutsouleris, N.; Gaser, C.; Arsic, M.; Weindl, A.; Reiser, M.; Meisenzahl, E.M. Variation within the Huntington’s Disease Gene Influences Normal Brain Structure. PLoS ONE 2012, 7, e29809. [Google Scholar] [CrossRef]
- Aziz, N.A.; Jurgens, C.K.; Landwehrmeyer, G.B.; Van Roon-Mom, W.M.C.; Van Ommen, G.J.B.; Stijnen, T.; Roos, R.A.C.; EHDN Registry Study Group. Normal and mutant HTT interact to affect clinical severity and progression in Huntington disease. Neurology 2009, 73, 1280–1285, Erratum in: Neurology 2009, 73, 1608. Erratum in: Neurology 2011, 76, 202. [Google Scholar] [CrossRef]
- Djousse, L.; Knowlton, B.; Hayden, M.; Almqvist, E.W.; Brinkman, R.; Ross, C.; Margolis, R.; Rosenblatt, A.; Durr, A.; Dode, C.; et al. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am. J. Med. Genet. A 2003, 119, 279–282. [Google Scholar] [CrossRef]
- Volpi, E.; Terenzi, F.; Bagnoli, S.; Latorraca, S.; Nacmias, B.; Sorbi, S.; Piacentini, S.; Ferrari, C. Late-onset Huntington disease: An Italian cohort. J. Clin. Neurosci. 2021, 86, 58–63. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Jia, Y.; Zhong, S.; Sun, Y.; Zhou, Z.; Zhang, Z.; Huang, L. Microstructural Abnormalities of Basal Ganglia and Thalamus in Bipolar and Unipolar Disorders: A Diffusion Kurtosis and Perfusion Imaging Study. Psychiatry Investig. 2017, 14, 471–482. [Google Scholar] [CrossRef]
- Aylward, E.H.; Roberts-Twillie, J.V.; Barta, P.E.; Kumar, A.J.; Harris, G.J.; Geer, M.; Peyser, C.E.; Pearlson, G.D. Basal ganglia volumes and white matter hyperintensities in patients with bipolar disorder. Am. J. Psychiatry 1994, 151, 687–693. [Google Scholar] [CrossRef]


{kind=link}
| Patients (n = 69) | |
|---|---|
| Age (SD) | 53.91 (10.2) |
| Age at disease onset (SD) | 34.81 (13.4) |
| Sex | |
| Male (%) | 27 (39.1) |
| Female (%) | 42 (60.9) |
| Education in years (SD) | 10.25 (3.1) |
| Symptoms at onset | |
| Maniacal (%) | 11 (15.9) |
| Depression (%) | 34 (49.2) |
| Mix (%) | 24 (34.7) |
| Type 1 bipolar disorder (%) | 44 (63.7) |
| Family history of psychiatric disorders (%) | 30 (43.5) |
| HTT gene * | |
| 17.7 (2.1) |
| 21.03 (3.3) |
| - |
| 0 |
| 7 (10.2) |
| No * IA Carriers (n = 62) | * IA Carriers (n = 7) | p | |
|---|---|---|---|
| Age at disease onset (SD) | 33.7 (13.5) | 43.3 (9.9) | 0.048 |
| Sex | |||
| Male (%) | 23 (37.1%) | 4 (57.3%) | 0.38 |
| Female (%) | 39 (62.9%) | 3 (42.8) | |
| Symptoms at onset | |||
| Maniacal (%) | 9 (14.5) | 2 (28.5) | 0.89 |
| Depression (%) | 30 (48.4) | 4 (57.1) | |
| Mix (%) | 23 (37.1) | 1 (14.2) | |
| Family history of psychiatric disorder (%) | 39 (45.1) | 2 (28.5) | 0.09 |
| CAG 1 (SD) | 17.37 (1.8) | 20.6 (2.3) | 0.09 |
| CAG 2 (SD) | 20.2 (2.08) | 28.6 (1.4) | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrari, C.; Capacci, E.; Bagnoli, S.; Ingannato, A.; Sorbi, S.; Nacmias, B. The Huntington’s Disease Gene in an Italian Cohort of Patients with Bipolar Disorder. Genes 2023, 14, 1681. https://doi.org/10.3390/genes14091681
Ferrari C, Capacci E, Bagnoli S, Ingannato A, Sorbi S, Nacmias B. The Huntington’s Disease Gene in an Italian Cohort of Patients with Bipolar Disorder. Genes. 2023; 14(9):1681. https://doi.org/10.3390/genes14091681
Chicago/Turabian StyleFerrari, Camilla, Elena Capacci, Silvia Bagnoli, Assunta Ingannato, Sandro Sorbi, and Benedetta Nacmias. 2023. "The Huntington’s Disease Gene in an Italian Cohort of Patients with Bipolar Disorder" Genes 14, no. 9: 1681. https://doi.org/10.3390/genes14091681
APA StyleFerrari, C., Capacci, E., Bagnoli, S., Ingannato, A., Sorbi, S., & Nacmias, B. (2023). The Huntington’s Disease Gene in an Italian Cohort of Patients with Bipolar Disorder. Genes, 14(9), 1681. https://doi.org/10.3390/genes14091681