
Heterozygosity of ALG9 in Association with Autosomal Dominant Polycystic Liver Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Selection
2.2. Genetic Screening
2.3. Conservation Analysis
2.4. 3D Modeling
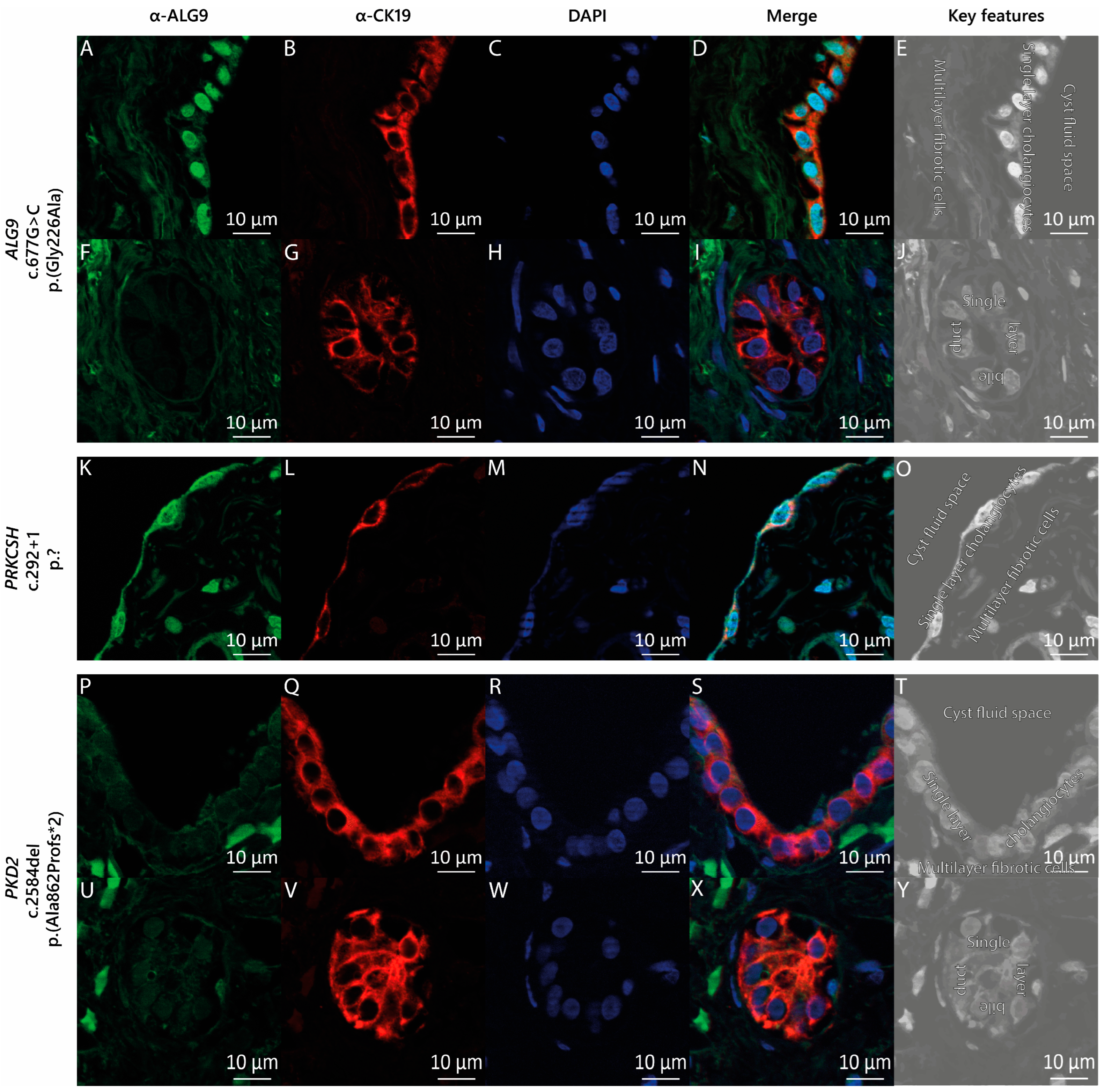
2.5. Fluorescent Immunohistochemistry
2.6. Ethics Approval
3. Results
3.1. Clinical Characteristics
3.2. Pathogenicity Prediction
3.3. ALG9 Expression in the Cyst Wall Lining
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohanty, S.; Chaudhary, B.P.; Zoetewey, D. Structural Insight into the Mechanism of N-Linked Glycosylation by Oligosaccharyltransferase. Biomolecules 2020, 10, 624. [Google Scholar] [CrossRef]
- Xu, C.; Ng, D.T.W. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 2015, 16, 742–752. [Google Scholar] [CrossRef]
- Aebi, M. N-linked protein glycosylation in the ER. Biochim. Biophys. Acta 2013, 1833, 2430–2437. [Google Scholar] [CrossRef] [PubMed]
- Vleugels, W.; Keldermans, L.; Jaeken, J.; Butters, T.D.; Michalski, J.C.; Matthijs, G.; Foulquier, F. Quality control of glycoproteins bearing truncated glycans in an ALG9-defective (CDG-IL) patient. Glycobiology 2009, 19, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Tham, E.; Eklund, E.A.; Hammarsjö, A.; Bengtson, P.; Geiberger, S.; Lagerstedt-Robinson, K.; Malmgren, H.; Nilsson, D.; Grigelionis, G.; Conner, P.; et al. A novel phenotype in N-glycosylation disorders: Gillessen-Kaesbach-Nishimura skeletal dysplasia due to pathogenic variants in ALG9. Eur. J. Hum. Genet. 2016, 24, 198–207. [Google Scholar] [CrossRef]
- Frank, C.G.; Eyaid, W.; Berger, E.G.; Aebi, M.; Grubenmann, C.E.; Hennet, T. Identification and Functional Analysis of a Defect in the Human ALG9 Gene: Definition of Congenital Disorder of Glycosylation Type IL. Am. J. Hum. Genet. 2004, 75, 146–150. [Google Scholar] [CrossRef]
- Weinstein, M.; Schollen, E.; Matthijs, G.; Neupert, C.; Hennet, T.; Grubenmann, C.E.; Frank, C.G.; Aebi, M.; Clarke, J.T.R.; Griffiths, A.; et al. CDG-IL: An infant with a novel mutation in the ALG9 gene and additional phenotypic features. Am. J. Med. Genet. Part A 2005, 136A, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Alsubhi, S.; Alhashem, A.; Faqeih, E.; Alfadhel, M.; Alfaifi, A.; Altuwaijri, W.; Alsahli, S.; Aldhalaan, H.; Alkuraya, F.S.; Hundallah, K. Congenital disorders of glycosylation: The Saudi experience. Am. J. Med. Genet. Part A 2017, 173, 2614–2621. [Google Scholar] [CrossRef]
- Davis, K.; Webster, D.; Smith, C.; Jackson, S.; Sinasac, D.; Seargeant, L.; Wei, X.-C.; Ferreira, P.; Midgley, J.; Foster, Y.; et al. ALG9-CDG: New clinical case and review of the literature. Mol. Genet. Metab. Rep. 2017, 13, 55–63. [Google Scholar] [CrossRef]
- Himmelreich, N.; Dimitrov, B.; Zielonka, M.; Hüllen, A.; Hoffmann, G.F.; Juenger, H.; Müller, H.; Lorenz, I.; Busse, B.; Marschall, C.; et al. Missense variant c.1460 T > C (p.L487P) enhances protein degradation of ER mannosyltransferase ALG9 in two new ALG9-CDG patients presenting with West syndrome and review of the literature. Mol. Genet. Metab. 2022, 136, 274–281. [Google Scholar] [CrossRef]
- Kaymak, D.; Alpay, V.; Davutoglu, E.A.; Elci, O.; Yigin, A.K.; Tuysuz, B.; Madazli, R. Gillessen-Kaesbach-Nishimura syndrome in two fetuses from Turkey. Am. J. Med. Genet. A 2023, 191, 617–623. [Google Scholar] [CrossRef]
- Besse, W.; Chang, A.R.; Luo, J.Z.; Triffo, W.J.; Moore, B.S.; Gulati, A.; Hartzel, D.N.; Mane, S.; Center, R.G.; Torres, V.E.; et al. ALG9 Mutation Carriers Develop Kidney and Liver Cysts. J. Am. Soc. Nephrol. 2019, 30, 2091–2102. [Google Scholar] [CrossRef]
- Schönauer, R.; Baatz, S.; Nemitz-Kliemchen, M.; Frank, V.; Petzold, F.; Sewerin, S.; Popp, B.; Münch, J.; Neuber, S.; Bergmann, C.; et al. Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genet. Med. 2020, 22, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Bae, K.T.; Zhu, F.; Chapman, A.B.; Torres, V.E.; Grantham, J.J.; Guay-Woodford, L.M.; Baumgarten, D.A.; King, B.F.J.; Wetzel, L.H.; Kenney, P.J.; et al. Magnetic Resonance Imaging Evaluation of Hepatic Cysts in Early Autosomal-Dominant Polycystic Kidney Disease: The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease Cohort. Clin. J. Am. Soc. Nephrol. 2006, 1, 64–69. [Google Scholar] [CrossRef]
- van Aerts, R.M.M.; van de Laarschot, L.F.M.; Banales, J.M.; Drenth, J.P.H. Clinical management of polycystic liver disease. J. Hepatol. 2018, 68, 827–837. [Google Scholar] [CrossRef]
- Masyuk, T.V.; Masyuk, A.I.; LaRusso, N.F. Polycystic Liver Disease: Advances in Understanding and Treatment. Annu. Rev. Pathol. 2022, 17, 251–269. [Google Scholar] [CrossRef]
- Drenth, J.; Barten, T.; Hartog, H.; Nevens, F.; Taubert, R.; Torra Balcells, R.; Vilgrain, V.; Böttler, T. EASL Clinical Practice Guidelines on the management of cystic liver diseases. J. Hepatol. 2022, 77, 1083–1108. [Google Scholar] [CrossRef]
- Gabow, P.A.; Johnson, A.M.; Kaehny, W.D.; Manco-Johnson, M.L.; Duley, I.T.; Everson, G.T. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology 1990, 11, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Neijenhuis, M.K.; Kievit, W.; Verheesen, S.M.; D'Agnolo, H.M.; Gevers, T.J.; Drenth, J.P. Impact of liver volume on polycystic liver disease-related symptoms and quality of life. United Eur. Gastroenterol. J. 2018, 6, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Duijzer, R.; Barten, T.R.M.; Staring, C.B.; Drenth, J.P.H.; Gevers, T.J.G. Treatment of Polycystic Liver Disease: Impact on Patient-reported Symptom Severity and Health-related Quality of Life. J. Clin. Gastroenterol. 2022, 56, 731–739. [Google Scholar] [CrossRef]
- Adam, R.; Karam, V.; Cailliez, V.; Grady, J.G.O.; Mirza, D.; Cherqui, D.; Klempnauer, J.; Salizzoni, M.; Pratschke, J.; Jamieson, N.; et al. 2018 Annual Report of the European Liver Transplant Registry (ELTR)—50-year evolution of liver transplantation. Transpl. Int. 2018, 31, 1293–1317. [Google Scholar] [CrossRef] [PubMed]
- Alsager, M.; Neong, S.F.; Gandhi, R.; Teriaky, A.; Tang, E.; Skaro, A.; Qumosani, K.; Lilly, L.; Galvin, Z.; Selzner, N.; et al. Liver transplantation in adult polycystic liver disease: The Ontario experience. BMC Gastroenterol. 2021, 21, 115. [Google Scholar] [CrossRef]
- Norcia, L.F.; Watanabe, E.M.; Hamamoto Filho, P.T.; Hasimoto, C.N.; Pelafsky, L.; de Oliveira, W.K.; Sassaki, L.Y. Polycystic Liver Disease: Pathophysiology, Diagnosis and Treatment. Hepat. Med. 2022, 14, 135–161. [Google Scholar] [CrossRef] [PubMed]
- Olaizola, P.; Rodrigues, P.M.; Caballero-Camino, F.J.; Izquierdo-Sanchez, L.; Aspichueta, P.; Bujanda, L.; Larusso, N.F.; Drenth, J.P.H.; Perugorria, M.J.; Banales, J.M. Genetics, pathobiology and therapeutic opportunities of polycystic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 585–604. [Google Scholar] [CrossRef]
- Boerrigter, M.M.; Bongers, E.; Lugtenberg, D.; Nevens, F.; Drenth, J.P.H. Polycystic liver disease genes: Practical considerations for genetic testing. Eur. J. Med. Genet. 2021, 64, 104160. [Google Scholar] [CrossRef]
- Lemoine, H.; Raud, L.; Foulquier, F.; Sayer, J.A.; Lambert, B.; Olinger, E.; Lefèvre, S.; Knebelmann, B.; Harris, P.C.; Trouvé, P.; et al. Monoallelic pathogenic ALG5 variants cause atypical polycystic kidney disease and interstitial fibrosis. Am. J. Hum. Genet. 2022, 109, 1484–1499. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef]
- Pisani, I.; Allinovi, M.; Palazzo, V.; Zanelli, P.; Gentile, M.; Farina, M.T.; Giuliotti, S.; Cravedi, P.; Delsante, M.; Maggiore, U.; et al. More dissimilarities than affinities between DNAJB11-PKD and ADPKD. Clin. Kidney J. 2022, 15, 1179–1187. [Google Scholar] [CrossRef]
- Huynh, V.T.; Audrézet, M.P.; Sayer, J.A.; Ong, A.C.; Lefevre, S.; Le Brun, V.; Després, A.; Senum, S.R.; Chebib, F.T.; Barroso-Gil, M.; et al. Clinical spectrum, prognosis and estimated prevalence of DNAJB11-kidney disease. Kidney Int. 2020, 98, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Senum, S.R.; Li, Y.S.M.; Benson, K.A.; Joli, G.; Olinger, E.; Lavu, S.; Madsen, C.D.; Gregory, A.V.; Neatu, R.; Kline, T.L.; et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am. J. Hum. Genet. 2022, 109, 136–156. [Google Scholar] [CrossRef]
- Wilson, E.M.; Choi, J.; Torres, V.E.; Somlo, S.; Besse, W. Large Deletions in GANAB and SEC63 Explain 2 Cases of Polycystic Kidney and Liver Disease. Kidney Int. Rep. 2020, 5, 727–731. [Google Scholar] [CrossRef]
- Besse, W.; Dong, K.; Choi, J.; Punia, S.; Fedeles, S.V.; Choi, M.; Gallagher, A.R.; Huang, E.B.; Gulati, A.; Knight, J.; et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Investig. 2017, 127, 1772–1785. [Google Scholar] [CrossRef] [PubMed]
- Apple, B.; Sartori, G.; Moore, B.; Chintam, K.; Singh, G.; Anand, P.M.; Strande, N.; Mirshahi, T.; Triffo, W.; Chang, A.R. Individuals heterozygous for ALG8 protein-truncating variants are at increased risk of a mild cystic kidney disease. Kidney Int. 2022, 103, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, M.M.; Te Morsche, R.H.M.; Venselaar, H.; Pastoors, N.; Geerts, A.M.; Hoorens, A.; Drenth, J.P.H. Novel α-1,3-Glucosyltransferase Variants and Their Broad Clinical Polycystic Liver Disease Spectrum. Genes 2023, 14, 1652. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, W.; Liu, J.; Liu, S.; Huang, W.; Shi, Y.; Zhang, X.; Lu, C.; Xie, W. A BBS4 mutation causes autosomal dominant polycystic liver disease. Genes Dis. 2023, 11, 72–75. [Google Scholar] [CrossRef]
- Wang, J.; Yang, H.; Guo, R.; Sang, X.; Mao, Y. Association of a novel PKHD1 mutation in a family with autosomal dominant polycystic liver disease. Ann. Transl. Med. 2021, 9, 120. [Google Scholar] [CrossRef] [PubMed]
- van de Laarschot, L.F.M.; Te Morsche, R.H.M.; Hoischen, A.; Venselaar, H.; Roelofs, H.M.; Cnossen, W.R.; Banales, J.M.; Roepman, R.; Drenth, J.P.H. Novel GANAB variants associated with polycystic liver disease. Orphanet. J. Rare Dis. 2020, 15, 302. [Google Scholar] [CrossRef] [PubMed]
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef]
- Delbarba, E.; Econimo, L.; Dordoni, C.; Martin, E.; Mazza, C.; Savoldi, G.; Alberici, F.; Scolari, F.; Izzi, C. Expanding the variability of the ADPKD-GANAB clinical phenotype in a family of Italian ancestry. J. Nephrol. 2022, 35, 645–652. [Google Scholar] [CrossRef]
- Besse, W.; Choi, J.; Ahram, D.; Mane, S.; Sanna-Cherchi, S.; Torres, V.; Somlo, S. A noncoding variant in GANAB explains isolated polycystic liver disease (PCLD) in a large family. Hum. Mutat. 2018, 39, 378–382. [Google Scholar] [CrossRef]
- Cnossen, W.R.; te Morsche, R.H.M.; Hoischen, A.; Gilissen, C.; Chrispijn, M.; Venselaar, H.; Mehdi, S.; Bergmann, C.; Veltman, J.A.; Drenth, J.P.H. Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 5343–5348. [Google Scholar] [CrossRef]
- Cnossen, W.R.; te Morsche, R.H.M.; Hoischen, A.; Gilissen, C.; Venselaar, H.; Mehdi, S.; Bergmann, C.; Losekoot, M.; Breuning, M.H.; Peters, D.J.M.; et al. LRP5 variants may contribute to ADPKD. Eur. J. Hum. Genet. 2016, 24, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Schlevogt, B.; Schlieper, V.; Krader, J.; Schröter, R.; Wagner, T.; Weiand, M.; Zibert, A.; Schmidt, H.H.; Bergmann, C.; Nedvetsky, P.I.; et al. A SEC61A1 variant is associated with autosomal dominant polycystic liver disease. Liver Int. 2023, 43, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Obaji, J.; Dupuis, A.; Paterson, A.D.; Magistroni, R.; Dicks, E.; Parfrey, P.; Cramer, B.; Coto, E.; Torra, R.; et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J. Am. Soc. Nephrol. 2009, 20, 205–212. [Google Scholar] [CrossRef] [PubMed]
- van de Laarschot, L.F.M.; te Morsche, R.H.M.; Roelofs, H.M.; Salomon, J.; Hoischen, A.; Nevens, F.; Peters, D.J.M.; Roepman, R.; Drenth, J.P.H. Molecular inversion probe analysis identifies five novel genes associated with polycystic liver disease. 2022.
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 1 December 2022).
- Boomsma, D.I.; Wijmenga, C.; Slagboom, E.P.; Swertz, M.A.; Karssen, L.C.; Abdellaoui, A.; Ye, K.; Guryev, V.; Vermaat, M.; van Dijk, F.; et al. The Genome of the Netherlands: Design, and project goals. Eur. J. Hum. Genet. 2014, 22, 221–227. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 27.20.21–27.20.41. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Heringa, J. Two strategies for sequence comparison: Profile-preprocessed and secondary structure-induced multiple alignment. Comput. Chem. 1999, 23, 341–364. [Google Scholar] [CrossRef] [PubMed]
- Venselaar, H.; Te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Janssen, M.J.; Waanders, E.; Te Morsche, R.H.; Xing, R.; Dijkman, H.B.; Woudenberg, J.; Drenth, J.P. Secondary, somatic mutations might promote cyst formation in patients with autosomal dominant polycystic liver disease. Gastroenterology 2011, 141, 2056–2063.e2. [Google Scholar] [CrossRef]
- Watnick, T.J.; Torres, V.E.; Gandolph, M.A.; Qian, F.; Onuchic, L.F.; Klinger, K.W.; Landes, G.; Germino, G.G. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol. Cell 1998, 2, 247–251. [Google Scholar] [CrossRef]
- Pei, Y.; Watnick, T.; He, N.; Wang, K.; Liang, Y.; Parfrey, P.; Germino, G.; St George-Hyslop, P. Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1999, 10, 1524–1529. [Google Scholar] [CrossRef]
- Janssen, M.J.; Salomon, J.; te Morsche, R.H.M.; Drenth, J.P.H. Loss of Heterozygosity Is Present in SEC63 Germline Carriers with Polycystic Liver Disease. PLoS ONE 2012, 7, e50324. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.J.; Salomon, J.; Cnossen, W.R.; Bergmann, C.; Pfundt, R.; Drenth, J.P. Somatic loss of polycystic disease genes contributes to the formation of isolated and polycystic liver cysts. Gut 2015, 64, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Wills, E.S.; Cnossen, W.R.; Veltman, J.A.; Woestenenk, R.; Steehouwer, M.; Salomon, J.; Te Morsche, R.H.; Huch, M.; Hehir-Kwa, J.Y.; Banning, M.J.; et al. Chromosomal abnormalities in hepatic cysts point to novel polycystic liver disease genes. Eur. J. Hum. Genet. 2016, 24, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Drenth, J.P.H.; te Morsche, R.H.M.; Smink, R.; Bonifacino, J.S.; Jansen, J.B.M.J. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 2003, 33, 345–347. [Google Scholar] [CrossRef]
- Li, A.; Davila, S.; Furu, L.; Qian, Q.; Tian, X.; Kamath, P.S.; King, B.F.; Torres, V.E.; Somlo, S. Mutations in PRKCSH Cause Isolated Autosomal Dominant Polycystic Liver Disease. Am. J. Hum. Genet. 2003, 72, 691–703. [Google Scholar] [CrossRef]
- Reynolds, D.M.; Falk, C.T.; Li, A.; King, B.F.; Kamath, P.S.; Huston, J.; Shub, C.; Iglesias, D.M.; Martin, R.S.; Pirson, Y.; et al. Identification of a Locus for Autosomal Dominant Polycystic Liver Disease, on Chromosome 19p13.2-13.1. Am. J. Hum. Genet. 2000, 67, 1598–1604. [Google Scholar] [CrossRef]
- Davila, S.; Furu, L.; Gharavi, A.G.; Tian, X.; Onoe, T.; Qian, Q.; Li, A.; Cai, Y.; Kamath, P.S.; King, B.F.; et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat. Genet. 2004, 36, 575–577. [Google Scholar] [CrossRef]
- Pirson, Y.; Lannoy, N.; Peters, D.; Geubel, A.; Gigot, J.F.; Breuning, M.; Verellen-Dumoulin, C. Isolated polycystic liver disease as a distinct genetic disease, unlinked to polycystic kidney disease 1 and polycystic kidney disease 2. Hepatology 1996, 23, 249–252. [Google Scholar] [CrossRef]
- Tannous, A.; Pisoni, G.B.; Hebert, D.N.; Molinari, M. N-linked sugar-regulated protein folding and quality control in the ER. Semin. Cell Dev. Biol. 2015, 41, 79–89. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boerrigter, M.M.; Duijzer, R.; te Morsche, R.H.M.; Drenth, J.P.H. Heterozygosity of ALG9 in Association with Autosomal Dominant Polycystic Liver Disease. Genes 2023, 14, 1755. https://doi.org/10.3390/genes14091755
Boerrigter MM, Duijzer R, te Morsche RHM, Drenth JPH. Heterozygosity of ALG9 in Association with Autosomal Dominant Polycystic Liver Disease. Genes. 2023; 14(9):1755. https://doi.org/10.3390/genes14091755
Chicago/Turabian StyleBoerrigter, Melissa M., Renée Duijzer, René H. M. te Morsche, and Joost P. H. Drenth. 2023. "Heterozygosity of ALG9 in Association with Autosomal Dominant Polycystic Liver Disease" Genes 14, no. 9: 1755. https://doi.org/10.3390/genes14091755
APA StyleBoerrigter, M. M., Duijzer, R., te Morsche, R. H. M., & Drenth, J. P. H. (2023). Heterozygosity of ALG9 in Association with Autosomal Dominant Polycystic Liver Disease. Genes, 14(9), 1755. https://doi.org/10.3390/genes14091755