
Polymorphisms and Pharmacogenomics of NQO2: The Past and the Future




Abstract
:1. Introduction
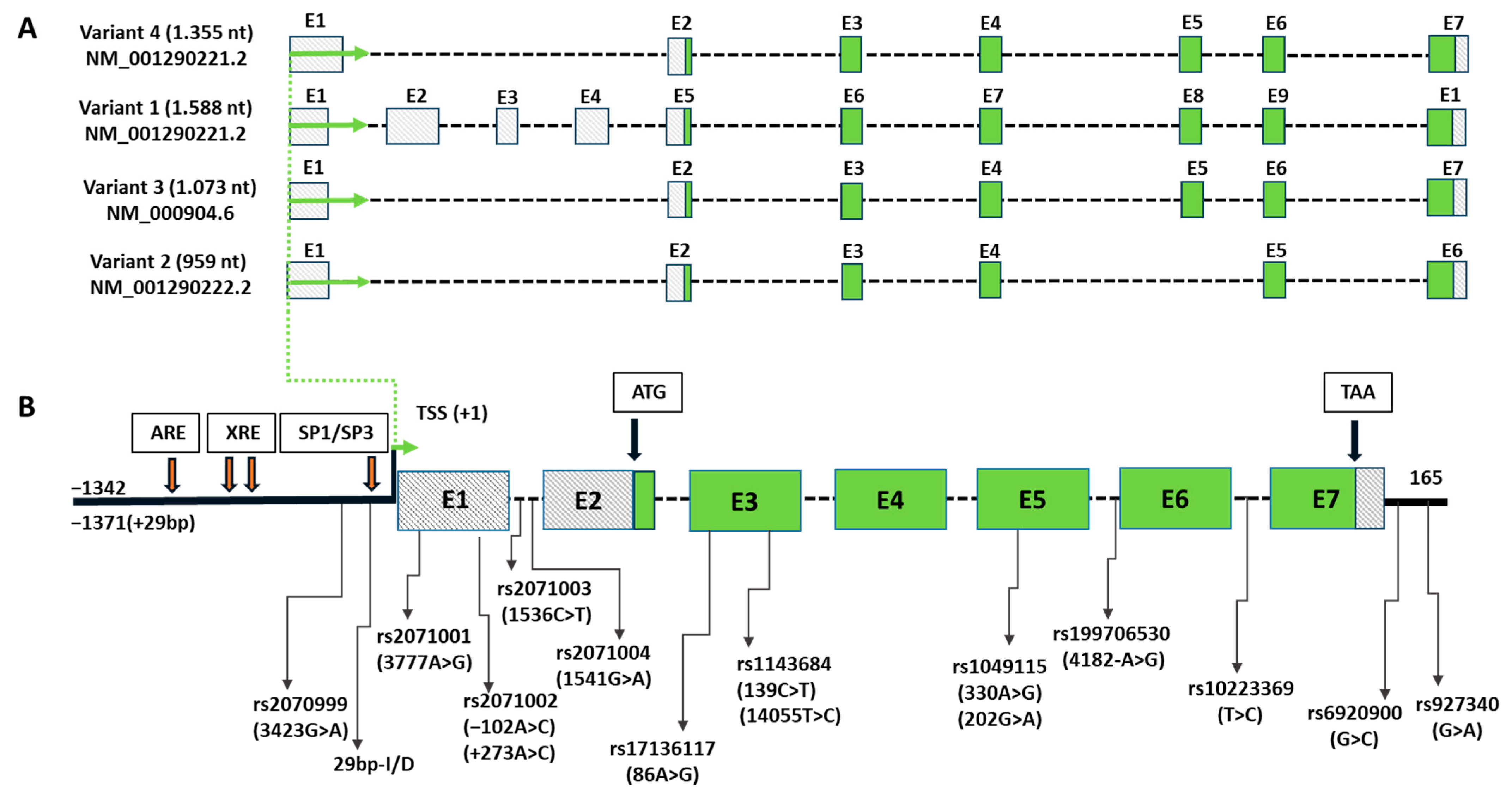
1.1. NQO2 Gene Structure and Its Polymorphic Variants
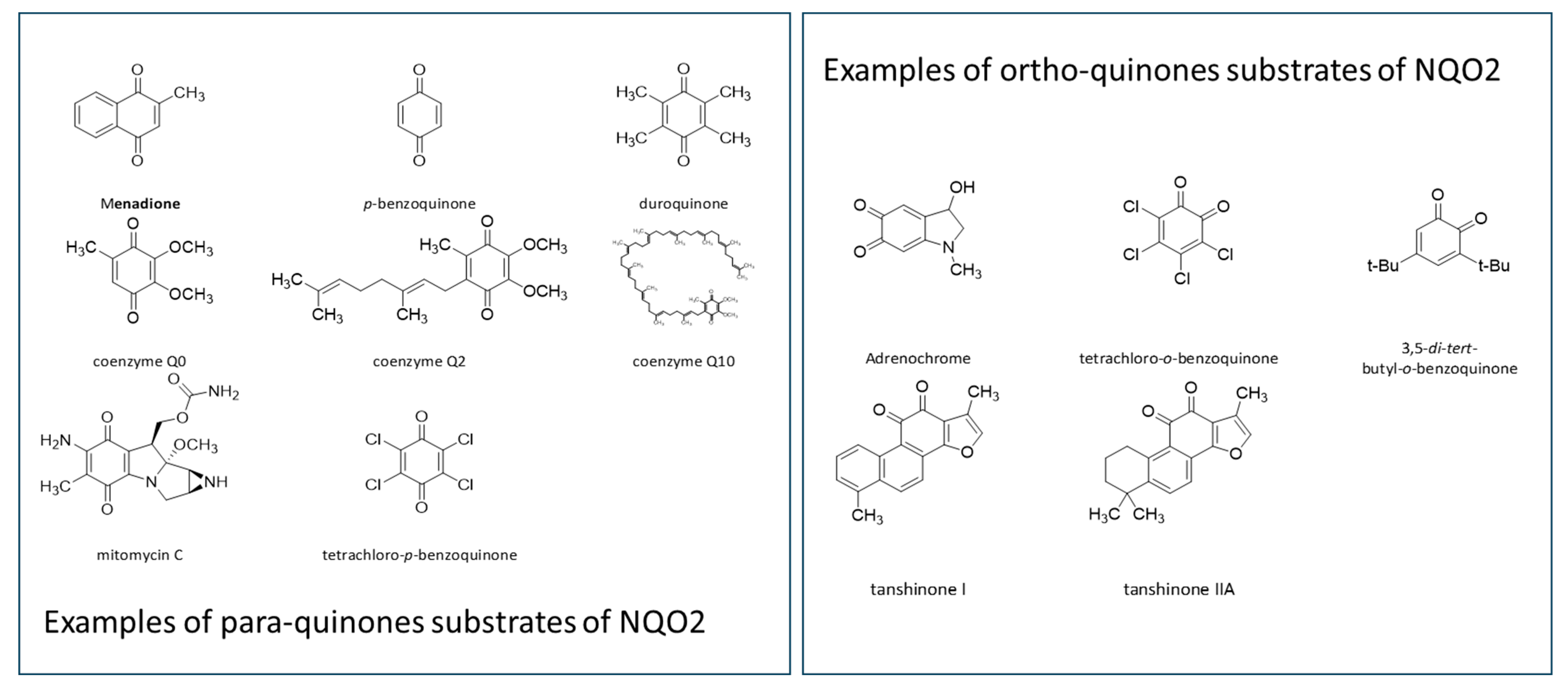
1.2. The Role of NQO2 in Drug Metabolism
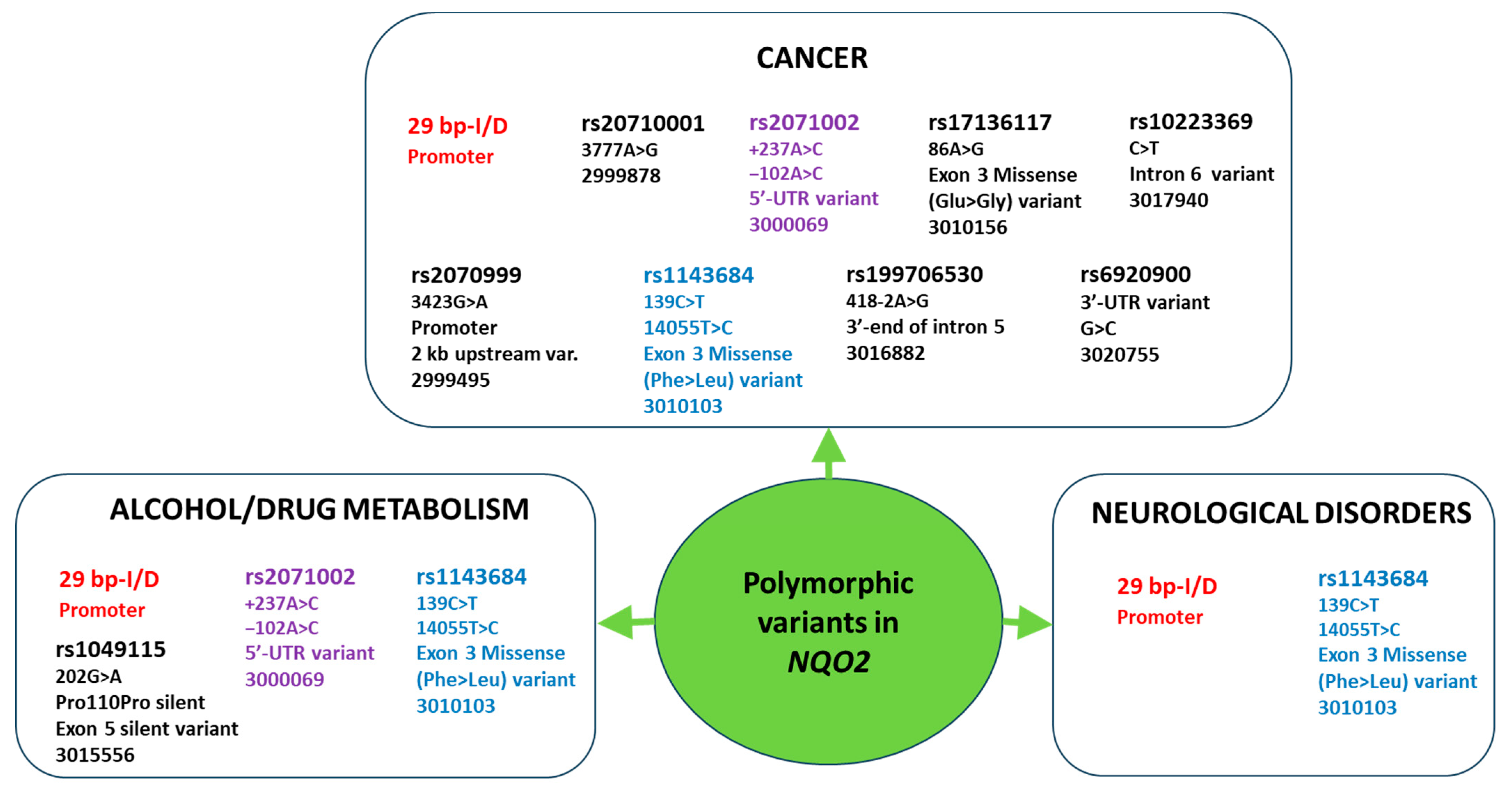
2. The Functional Role of NQO2 Polymorphic Variants in Pathology
2.1. Cancer
2.1.1. Breast Cancer (BC)

{kind=link}
{kind=link}
{kind=link}
| Polymorphism Names * and Location | Allele | Possible Molecular Function | Ref. | Pathology | Higher Risk Allele | Clinical Reference |
|---|---|---|---|---|---|---|
| 29 bp-I/D Promoter | D | No Sp3 site Higher NQO2 expression and activity | [16,17,18,48] | Breast cancer | I | [18] |
| Parkinson’s Disease Idiopathic | D | [49] | ||||
| Schizophrenia | D | [50] | ||||
| Methamphetamine-related psychosis | D | [51] | ||||
| Alcoholism and alcohol withdrawal symptoms | D | [52] | ||||
| I | Sp3 site present Lower NQO2 expression and activity | Early breast cancer | I | [53] | ||
| Chronic Alcoholic pancreatitis | No | [54] | ||||
| Primary Breast cancer | I | [48] | ||||
| Parkinson’s Disease Idiopathic | No | [55] | ||||
| Papillary thyroid microcarcinoma (PTMC) | I | [56] | ||||
| rs2070999 3423 G>A Promoter 2 kb upstream var. 2999495 | A | Lower NQO2 activity | [19,57] | Esophageal cancer (EC) | A | [57] |
| Esophageal cancer (EC) | No | [58] | ||||
| G | Higher NQO2 activity | Gastric cancer | No | [59] | ||
| Bladder cancer | A | [19] | ||||
| Prostate cancer | No | [20] | ||||
| rs20710001 3777 A>G 2999878 | A | Higher NQO2 activity | [2,19,21] | Bladder cancer | G | [19] |
| G | Lower NQO2 activity | Ovarian cancer | ||||
| rs2071002 +237 A>C −102 A>C 1507 C>A 5′-UTR variant 3000069 | A | Abolishes Sp1 site Lower NQO2 expression | [18,48] | Breast cancer | A | [18] |
| Primary breast cancer | C | [48] | ||||
| C | Sp1 site present Higher NQO2 expression | Sporadic breast cancer | C | [60] | ||
| Epithelial ovarian cancer | A | [61] | ||||
| rs2071003 1536 C>T Intron 1 variant 3000098 | T | No MZF-1 binding site Low NQO2 expression | [62] | Clozapine-induced Agranulocytosis | T | [62] |
| C | MZF-1 binding site High NQO2 expression | |||||
| rs2071004 1541 G>A Intron 1 variant 3000103 | A | Low NQO2 mRNA | [63] | Agranulocytosis | A | [63] |
| G | High NQO2 mRNA | |||||
| rs17136117 86 A>G Exon 3 Missense (Glu>Gly) variant 3010156 | A | Unknown | Sporadic Breast cancer | G | [60] | |
| G | ||||||
| rs1143684 139 C>T 14055 T>C 372 T>C Exon 3 Missense (Phe>Leu) variant 3010103 | T (Phe) | Higher NQO2 activity | [19,21,48,62] | Primary Breast cancer | T | [48] |
| Breast cancer | No | [64] | ||||
| Localized Prostate cancer | C | [65] | ||||
| C (Leu) | Lower NQO2 activity | Pancreatic cancer | No | [66] | ||
| Bladder cancer | No | [67] | ||||
| Bladder cancer | C | [19] | ||||
| Ovarian cancer | C | [19] | ||||
| Cognitive decline | C | [68] | ||||
| Clozapine induced- Agranulocytosis | C | [62] | ||||
| rs1049115 202 G>A Pro110Pro silent Exon 5 3015556 | A | Silent mutation | [62] | Clozapine induced- Agranulocytosis | A | [62] |
| G | ||||||
| rs199706530 418−2 A>G 3′-end of intron 5 3016882 | A | Normal splicing | [69] | Hereditary Breast and Ovarian Cancer | G | [69] |
| G | Aberrant splicing: no acceptor splice site, truncated transcripts w/o exons 5 and 6 | |||||
| rs10223369 C>T Intron 6 variant 3017940 | T | Unknown | Localized Prostate cancer | T | [65] | |
| C | ||||||
| rs6920900 G>C 3′-UTR variant 3020755 | C | Unknown | Localized Prostate cancer | C | [65] | |
| G | ||||||
| rs927340 G>A 3′-UTR 3023484 | A | Unknown | Ovarian cancer | No | [70] | |
| G |
2.1.2. Other Cancers
2.2. Neurodegeneration
2.3. Memory and Brain Pathophysiology
3. NQO2 and Pharmacogenomics
3.1. Effects of NQO2 Polymorphisms on Cancer Therapy
3.2. Effects of NQO2 Polymorphisms on Other Drug Metabolism
4. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Janda, E.; Nepveu, F.; Calamini, B.; Ferry, G.; Boutin, J.A. Molecular Pharmacology of NRH:Quinone Oxidoreductase 2: A Detoxifying Enzyme Acting as an Undercover Toxifying Enzyme. Mol. Pharmacol. 2020, 98, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Babu, D.; Siraki, A.G. Interactions of the antioxidant enzymes NAD(P)H: Quinone oxidoreductase 1 (NQO1) and NRH: Quinone oxidoreductase 2 (NQO2) with pharmacological agents, endogenous biochemicals and environmental contaminants. Chem.-Biol. Interact. 2021, 345, 109574. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, N.; Zhang, G.; Sauve, A.A. NRH salvage and conversion to NAD+ requires NRH kinase activity by adenosine kinase. Nat. Metab. 2020, 2, 364–379. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Leung, K.K.; Walker, M.D.; Al Massri, S.; Shilton, B.H. The Unusual Cosubstrate Specificity of NQO2: Conservation Throughout the Amniotes and Implications for Cellular Function. Front. Pharmacol. 2022, 13, 838500. [Google Scholar] [CrossRef]
- Janda, E.; Martino, C.; Riillo, C.; Parafati, M.; Lascala, A.; Mollace, V.; Boutin, J.A. Apigenin and Luteolin Regulate Autophagy by Targeting NRH-Quinone Oxidoreductase 2 in Liver Cells. Antioxidants 2021, 10, 776. [Google Scholar] [CrossRef]
- Reinhardt, C.R.; Hu, Q.H.; Bresnahan, C.G.; Hati, S.; Bhattacharyya, S. Cyclic Changes in Active Site Polarization and Dynamics Drive the ‘Ping-pong’ Kinetics in NRH:Quinone Oxidoreductase 2: An Insight from QM/MM Simulations. ACS Catal. 2018, 8, 12015–12029. [Google Scholar] [CrossRef]
- Zhao, Q.; Yang, X.L.; Holtzclaw, W.D.; Talalay, P. Unexpected genetic and structural relationships of a long-forgotten flavoenzyme to NAD(P)H:quinone reductase (DT-diaphorase). Proc. Natl. Acad. Sci. USA 1997, 94, 1669–1674. [Google Scholar] [CrossRef]
- Bock, K.W. Ah receptor- and Nrf2-gene battery members: Modulators of quinone-mediated oxidative and endoplasmic reticulum stress. Biochem. Pharmacol. 2012, 83, 833–838. [Google Scholar] [CrossRef]
- Chhour, M.; Perio, P.; Gayon, R.; Ternet-Fontebasso, H.; Ferry, G.; Nepveu, F.; Boutin, J.A.; Sudor, J.; Reybier, K. Association of NQO2 With UDP-Glucuronosyltransferases Reduces Menadione Toxicity in Neuroblastoma Cells. Front. Pharmacol. 2021, 12, 660641. [Google Scholar] [CrossRef]
- Reybier, K.; Perio, P.; Ferry, G.; Bouajila, J.; Delagrange, P.; Boutin, J.A.; Nepveu, F. Insights into the redox cycle of human quinone reductase 2. Free Radic. Res. 2011, 45, 1184–1195. [Google Scholar] [CrossRef]
- Janda, E.; Parafati, M.; Aprigliano, S.; Carresi, C.; Visalli, V.; Sacco, I.; Ventrice, D.; Mega, T.; Vadalá, N.; Rinaldi, S.; et al. The antidote effect of quinone oxidoreductase 2 inhibitor against paraquat-induced toxicity in vitro and in vivo. Br. J. Pharmacol. 2013, 168, 46–59. [Google Scholar] [CrossRef]
- Janda, E.; Lascala, A.; Carresi, C.; Parafati, M.; Aprigliano, S.; Russo, V.; Savoia, C.; Ziviani, E.; Musolino, V.; Morani, F.; et al. Parkinsonian toxin-induced oxidative stress inhibits basal autophagy in astrocytes via NQO2/quinone oxidoreductase 2: Implications for neuroprotection. Autophagy 2015, 11, 1063–1080. [Google Scholar] [CrossRef] [PubMed]
- Janda, E.; Parafati, M.; Martino, C.; Crupi, F.; William, J.N.G.; Reybier, K.; Arbitrio, M.; Mollace, V.; Boutin, J.A. Autophagy and neuroprotection in astrocytes exposed to 6-hydroxydopamine is negatively regulated by NQO2: Relevance to parkinson’s disease. Sci. Rep. 2023, 13, 21624. [Google Scholar] [CrossRef] [PubMed]
- Vallucci, M.; Boutin, J.A.; Janda, E.; Blandel, F.; Musgrove, R.; Di Monte, D.; Ferry, G.; Michel, P.P.; Hirsch, E.C. The specific NQO2 inhibitor, S29434, only marginally improves the survival of dopamine neurons in MPTP-intoxicated mice. J. Neural Transm. 2023, 131, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dodson, A.; Mi, K.; Russo, D.P.; Scott, C.; Saulnier, M.; Snyder, K.; Racz, R. Aggregation and analysis of secondary pharmacology data from investigational new drug submissions at the US Food and Drug Administration. J. Pharmacol. Toxicol. Methods 2021, 111, 107098. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jaiswal, A.K. Sp3 repression of polymorphic human NRH:quinone oxidoreductase 2 gene promoter. Free Radic. Biol. Med. 2004, 37, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Le, W.-D.; Pan, T.; Stringer, J.L.; Jaiswal, A.K. Association of NRH:quinone oxidoreductase 2 gene promoter polymorphism with higher gene expression and increased susceptibility to Parkinson’s disease. J. Gerontol. A 2008, 63, 127–134. [Google Scholar] [CrossRef]
- Yu, K.-D.; Di, G.-H.; Yuan, W.-T.; Fan, L.; Wu, J.; Hu, Z.; Shen, Z.-Z.; Zheng, Y.; Huang, W.; Shao, Z.-M. Functional polymorphisms, altered gene expression and genetic association link NRH:quinone oxidoreductase 2 to breast cancer with wild-type p53. Hum. Mol. Genet. 2009, 18, 2502–2517. [Google Scholar] [CrossRef]
- Jamieson, D.; Wilson, K.; Pridgeon, S.; Margetts, J.P.; Edmondson, R.J.; Leung, H.Y.; Knox, R.; Boddy, A.V. NAD(P)H:quinone oxidoreductase 1 and nrh:quinone oxidoreductase 2 activity and expression in bladder and ovarian cancer and lower NRH:quinone oxidoreductase 2 activity associated with an NQO2 exon 3 single-nucleotide polymorphism. Clin. Cancer Res. 2007, 13, 1584–1590. [Google Scholar] [CrossRef]
- Mandal, R.K.; Nissar, K.; Mittal, R.D. Genetic variants in metabolizing genes NQO1, NQO2, MTHFR and risk of prostate cancer: A study from North India. Mol. Biol. Rep. 2012, 39, 11145–11152. [Google Scholar] [CrossRef]
- Megarity, C.F.; Gill, J.R.; Caraher, M.C.; Stratford, I.J.; Nolan, K.A.; Timson, D.J. The two common polymorphic forms of human NRH-quinone oxidoreductase 2 (NQO2) have different biochemical properties. FEBS Lett. 2014, 588, 1666–1672. [Google Scholar] [CrossRef]
- Testa, B.; Krämer, S.D. The biochemistry of drug metabolism—An introduction: Part 1. Principles and overview. Chem. Biodivers. 2006, 3, 1053–1101. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Krämer, S.D. The biochemistry of drug metabolism—An introduction: Part 2. Redox reactions and their enzymes. Chem. Biodivers. 2007, 4, 257–405. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Krämer, S.D. The biochemistry of drug metabolism—An introduction: Part 3. Reactions of hydrolysis and their enzymes. Chem. Biodivers. 2007, 4, 2031–2122. [Google Scholar] [CrossRef] [PubMed]
- Boutin, J.A. Camphoroquinone reduction: Another reaction catalyzed by rat liver cytosol 3 α-hydroxysteroid dehydrogenase. Biochim. Biophys. Acta 1986, 870, 463–472. [Google Scholar] [CrossRef]
- Bianchet, M.A.; Erdemli, S.B.; Amzel, L.M. Structure, function, and mechanism of cytosolic quinone reductases. Vitam. Horm. 2008, 78, 63–84. [Google Scholar] [CrossRef]
- Ross, D. Quinone reductases multitasking in the metabolic world. Drug Metab. Rev. 2004, 36, 639–654. [Google Scholar] [CrossRef]
- Rosenfeld, M.A.; Vasilyeva, A.D.; Yurina, L.V.; Bychkova, A.V. Oxidation of proteins: Is it a programmed process? Free Radic. Res. 2018, 52, 14–38. [Google Scholar] [CrossRef]
- Gaikwad, N.W.; Rogan, E.G.; Cavalieri, E.L. Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen ortho-quinones. Free Radic. Biol. Med. 2007, 43, 1289–1298. [Google Scholar] [CrossRef]
- Chhour, M.; Aubouy, A.; Bourgeade-Delmas, S.; Perio, P.; Ternet-Fontebasso, H.; Haidara, M.; Ferry, G.; Nepveu, F.; Boutin, J.A.; Reybier, K. Antimalarial Properties of Dunnione Derivatives as NQO2 Substrates. Molecules 2019, 24, 3697. [Google Scholar] [CrossRef]
- Ma, T.; Zhao, Q.; Wang, J.; Pan, Z.; Chen, J. A Sulfur Heterocyclic Quinone Cathode and a Multifunctional Binder for a High-Performance Rechargeable Lithium-Ion Battery. Angew. Chem. Int. Ed. 2016, 55, 6428–6432. [Google Scholar] [CrossRef] [PubMed]
- Hayat, F.; Sonavane, M.; Makarov, M.V.; Trammell, S.A.J.; McPherson, P.; Gassman, N.R.; Migaud, M.E. The Biochemical Pathways of Nicotinamide-Derived Pyridones. Int. J. Mol. Sci. 2021, 22, 1145. [Google Scholar] [CrossRef]
- Ciarlo, E.; Joffraud, M.; Hayat, F.; Giner, M.P.; Giroud-Gerbetant, J.; Sanchez-Garcia, J.L.; Rumpler, M.; Moco, S.; Migaud, M.E.; Cantó, C. Nicotinamide Riboside and Dihydronicotinic Acid Riboside Synergistically Increase Intracellular NAD+ by Generating Dihydronicotinamide Riboside. Nutrients 2022, 14, 2752. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Pérez, R.; Tammaro, A.; Schomakers, B.V.; Scantlebery, A.M.L.; Denis, S.; Elfrink, H.L.; Giroud-Gerbetant, J.; Cantó, C.; López-Leonardo, C.; McIntyre, R.L.; et al. Reduced nicotinamide mononucleotide is a new and potent NAD+ precursor in mammalian cells and mice. FASEB J. 2021, 35, e21456. [Google Scholar] [CrossRef] [PubMed]
- Biţă, A.; Scorei, I.R.; Ciocîlteu, M.V.; Nicolaescu, O.E.; Pîrvu, A.S.; Bejenaru, L.E.; Rău, G.; Bejenaru, C.; Radu, A.; Neamţu, J.; et al. Nicotinamide Riboside, a Promising Vitamin B3 Derivative for Healthy Aging and Longevity: Current Research and Perspectives. Molecules 2023, 28, 6078. [Google Scholar] [CrossRef] [PubMed]
- Vreones, M.; Mustapic, M.; Moaddel, R.; Pucha, K.A.; Lovett, J.; Seals, D.R.; Kapogiannis, D.; Martens, C.R. Oral nicotinamide riboside raises NAD+ and lowers biomarkers of neurodegenerative pathology in plasma extracellular vesicles enriched for neuronal origin. Aging Cell 2023, 22, e13754. [Google Scholar] [CrossRef]
- Cassagnes, L.-E.; Perio, P.; Ferry, G.; Moulharat, N.; Antoine, M.; Gayon, R.; Boutin, J.A.; Nepveu, F.; Reybier, K. In cellulo monitoring of quinone reductase activity and reactive oxygen species production during the redox cycling of 1,2 and 1,4 quinones. Free Radic. Biol. Med. 2015, 89, 126–134. [Google Scholar] [CrossRef]
- Boutin, J.A.; Ferry, G.; Reybier, K. A hypothesis on the equilibrium between dopamine toxicity and detoxification: The roles of NQO2 and UDP-glucuronosyltransferases. Gene Protein Dis. 2023, 2, 227. [Google Scholar] [CrossRef]
- Long, D.J., II; Iskander, K.; Gaikwad, A.; Arin, M.; Roop, D.R.; Knox, R.; Barrios, R.; Jaiswal, A.K. Disruption of dihydronicotinamide riboside:quinone oxidoreductase 2 (NQO2) leads to myeloid hyperplasia of bone marrow and decreased sensitivity to menadione toxicity. J. Biol. Chem. 2002, 277, 46131–46139. [Google Scholar] [CrossRef]
- Celli, C.M.; Tran, N.; Knox, R.; Jaiswal, A.K. NRH:quinone oxidoreductase 2 (NQO2) catalyzes metabolic activation of quinones and anti-tumor drugs. Biochem. Pharmacol. 2006, 72, 366–376. [Google Scholar] [CrossRef]
- Ran, L.Y.; Xiang, J.; Zeng, X.X.; He, W.W.; Dong, Y.T.; Yu, W.F.; Qi, X.L.; Xiao, Y.; Cao, K.; Zou, J.; et al. The influence of NQO2 on the dysfunctional autophagy and oxidative stress induced in the hippocampus of rats and in SH-SY5Y cells by fluoride. CNS Neurosci. Ther. 2023, 29, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Björklund, M. NQO2 is a reactive oxygen species generating off-target for acetaminophen. Mol. Pharm. 2014, 11, 4395–4404. [Google Scholar] [CrossRef] [PubMed]
- Cassagnes, L.-E.; Chhour, M.; Pério, P.; Sudor, J.; Gayon, R.; Ferry, G.; Boutin, J.A.; Nepveu, F.; Reybier, K. Oxidative stress and neurodegeneration: The possible contribution of quinone reductase 2. Free Radic. Biol. Med. 2018, 120, 56–61. [Google Scholar] [CrossRef]
- Cadenas, E. Antioxidant and prooxidant functions of DT-diaphorase in quinone metabolism. Biochem. Pharmacol. 1995, 49, 127–140. [Google Scholar] [CrossRef]
- Fu, Y.; Buryanovskyy, L.; Zhang, Z. Quinone reductase 2 is a catechol quinone reductase. J. Biol. Chem. 2008, 283, 23829–23835. [Google Scholar] [CrossRef] [PubMed]
- den Braver-Sewradj, S.P.; den Braver, M.W.; van Dijk, M.; Zhang, Y.; Dekker, S.J.; Wijaya, L.; Vermeulen, N.P.E.; Richert, L.; Commandeur, J.N.M.; Vos, J.C. Inter-individual Variability in Activity of the Major Drug Metabolizing Enzymes in Liver Homogenates of 20 Individuals. Curr. Drug Metab. 2018, 19, 370–381. [Google Scholar] [CrossRef] [PubMed]
- den Braver-Sewradj, S.P.; den Braver, M.W.; Toorneman, R.M.; van Leeuwen, S.; Zhang, Y.; Dekker, S.J.; Vermeulen, N.P.E.; Commandeur, J.N.M.; Vos, J.C. Reduction and Scavenging of Chemically Reactive Drug Metabolites by NAD(P)H:Quinone Oxidoreductase 1 and NRH:Quinone Oxidoreductase 2 and Variability in Hepatic Concentrations. Chem. Res. Toxicol. 2018, 31, 116–126. [Google Scholar] [CrossRef]
- Yu, K.-D.; Huang, A.-J.; Fan, L.; Li, W.-F.; Shao, Z.-M. Genetic variants in oxidative stress-related genes predict chemoresistance in primary breast cancer: A prospective observational study and validation. Cancer Res. 2012, 72, 408–419. [Google Scholar] [CrossRef]
- Harada, S.; Fujii, C.; Hayashi, A.; Ohkoshi, N. An association between idiopathic Parkinson’s disease and polymorphisms of phase II detoxification enzymes: Glutathione S-transferase M1 and quinone oxidoreductase 1 and 2. Biochem. Biophys. Res. Commun. 2001, 288, 887–892. [Google Scholar] [CrossRef]
- Harada, S.; Tachikawa, H.; Kawanishi, Y. A possible association between an insertion/deletion polymorphism of the NQO2 gene and schizophrenia. Psychiatr. Genet. 2003, 13, 205–209. [Google Scholar] [CrossRef]
- Ohgake, S.; Hashimoto, K.; Shimizu, E.; Koizumi, H.; Okamura, N.; Koike, K.; Matsuzawa, D.; Sekine, Y.; Inada, T.; Ozaki, N.; et al. Functional polymorphism of the NQO2 gene is associated with methamphetamine psychosis. Addict. Biol. 2005, 10, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Okubo, T.; Harada, S.; Higuchi, S.; Matsushita, S. Association analyses between polymorphisms of the phase II detoxification enzymes (GSTM1, NQO1, NQO2) and alcohol withdrawal symptoms. Alcohol Clin. Exp. Res. 2003, 27, 68S–71S. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, D.; Cresti, N.; Bray, J.; Sludden, J.; Griffin, M.J.; Hawsawi, N.M.; Famie, E.; Mould, E.V.; Verrill, M.W.; May, F.E.; et al. Two minor NQO1 and NQO2 alleles predict poor response of breast cancer patients to adjuvant doxorubicin and cyclophosphamide therapy. Pharmacogenet. Genom. 2011, 21, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Harada, S.; Yokoyama, A.; Naruse, S.; Hirota, M.; Nishimori, I.; Otsuki, M. Association analysis among polymorphisms of the various genes and chronic alcoholic pancreatitis. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. S1), S69–S72. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Farin, F.M.; Stapleton, P.; Viernes, H.; Quigley, S.D.; Powers, K.M.; Smith-Weller, T.; Franklin, G.M.; Longstreth, W.T.; Swanson, P.D.; et al. No associations between Parkinson’s disease and polymorphisms of the quinone oxidoreductase (NQO1, NQO2) genes. Neurosci. Lett. 2005, 375, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, K.S.; Lee, M.H.; Kim, Y.S.; Lee, M.H.; Lee, S.E.; Kim, Y.K.; Ryu, M.J.; Kim, S.J.; Choi, M.J.; et al. NAD(P)H: Quinone oxidoreductase 1 and NRH:quinone oxidoreductase 2 polymorphisms in papillary thyroid microcarcinoma: Correlation with phenotype. Yonsei Med. J. 2013, 54, 1158–1167. [Google Scholar] [CrossRef]
- Malik, M.A.; Zargar, S.A.; Mittal, B. Role of NQO1 609C>T and NQO2 -3423G>A gene polymorphisms in esophageal cancer risk in Kashmir valley and meta analysis. Mol. Biol. Rep. 2012, 39, 9095–9104. [Google Scholar] [CrossRef]
- Umar, M.; Upadhyay, R.; Kumar, S.; Ghoshal, U.C.; Mittal, B. Null association of NQO1 609C>T and NQO2 -3423G>A polymorphisms with susceptibility and prognosis of Esophageal cancer in north Indian population and meta-analysis. Cancer Epidemiol. 2012, 36, e373–e379. [Google Scholar] [CrossRef]
- Malik, M.A.; Zargar, S.A.; Mittal, B. Role of NQO1 609C>T and NQO2-3423G>A polymorphisms in susceptibility to gastric cancer in Kashmir valley. DNA Cell Biol. 2011, 30, 297–303. [Google Scholar] [CrossRef]
- Sirisena, N.D.; Adeyemo, A.; Kuruppu, A.I.; Samaranayake, N.; Dissanayake, V.H.W. Genetic Variants Associated with Clinicopathological Profiles in Sporadic Breast Cancer in Sri Lankan Women. J. Breast Cancer 2018, 21, 165–172. [Google Scholar] [CrossRef]
- Carron, J.; Brito, A.B.C.; Torelli, A.C.M.; Oliveira, C.; Derchain, S.F.M.; Lima, C.S.P.; Lourenço, G.J. Association between polymorphisms in xenobiotic detoxification-related genes with prognosis of epithelial ovarian cancer. Med. Oncol. 2016, 33, 112. [Google Scholar] [CrossRef] [PubMed]
- Ostrousky, O.; Meged, S.; Loewenthal, R.; Valevski, A.; Weizman, A.; Carp, H.; Gazit, E. NQO2 gene is associated with clozapine-induced agranulocytosis. Tissue Antigens 2003, 62, 483–491. [Google Scholar] [CrossRef] [PubMed]
- van der Weide, K.; Loovers, H.; Pondman, K.; Bogers, J.; van der Straaten, T.; Langemeijer, E.; Cohen, D.; Commandeur, J.; van der Weide, J. Genetic risk factors for clozapine-induced neutropenia and agranulocytosis in a Dutch psychiatric population. Pharmacogenom. J. 2017, 17, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-Y.; Barlow, W.E.; Albain, K.S.; Hong, C.-C.; Blanco, J.G.; Livingston, R.B.; Davis, W.; Rae, J.M.; Yeh, I.-T.; Hutchins, L.F.; et al. Nitric oxide synthase variants and disease-free survival among treated and untreated breast cancer patients in a Southwest Oncology Group clinical trial. Clin. Cancer Res. 2009, 15, 5258–5266. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, E.; Laverdière, I.; Audet-Walsh, E.; Caron, P.; Rouleau, M.; Fradet, Y.; Lacombe, L.; Guillemette, C. Steroidogenic germline polymorphism predictors of prostate cancer progression in the estradiol pathway. Clin. Cancer Res. 2014, 20, 2971–2983. [Google Scholar] [CrossRef] [PubMed]
- Mohelnikova-Duchonova, B.; Marsakova, L.; Vrana, D.; Holcatova, I.; Ryska, M.; Smerhovsky, Z.; Slamova, A.; Schejbalova, M.; Soucek, P. Superoxide dismutase and nicotinamide adenine dinucleotide phosphate: Quinone oxidoreductase polymorphisms and pancreatic cancer risk. Pancreas 2011, 40, 72–78. [Google Scholar] [CrossRef]
- Wen, H.; Ding, Q.; Fang, Z.-J.; Xia, G.-W.; Fang, J. Population study of genetic polymorphisms and superficial bladder cancer risk in Han-Chinese smokers in Shanghai. Int. Urol. Nephrol. 2009, 41, 855–864. [Google Scholar] [CrossRef]
- Payton, A.; Miyajima, F.; Ollier, W.; Rabbitt, P.; Pickles, A.; Weiss, V.; Pendleton, N.; Horan, M. Investigation of a functional quinine oxidoreductase (NQO2) polymorphism and cognitive decline. Neurobiol. Aging 2010, 31, 351–352. [Google Scholar] [CrossRef]
- Doddato, G.; Valentino, F.; Giliberti, A.; Papa, F.T.; Tita, R.; Bruno, L.P.; Resciniti, S.; Fallerini, C.; Benetti, E.; Palmieri, M.; et al. Exome sequencing in BRCA1-2 candidate familias: The contribution of other cancer susceptibility genes. Front. Oncol. 2021, 11, 649435. [Google Scholar] [CrossRef]
- Goode, E.L.; White, K.L.; Vierkant, R.A.; Phelan, C.M.; Cunningham, J.M.; Schildkraut, J.M.; Berchuck, A.; Larson, M.C.; Fridley, B.L.; Olson, J.E.; et al. Xenobiotic-Metabolizing gene polymorphisms and ovarian cancer risk. Mol. Carcinog. 2011, 50, 397–402. [Google Scholar] [CrossRef]
- Kaur, R.P.; Vasudeva, K.; Kumar, R.; Munshi, A. Role of p53 Gene in Breast Cancer: Focus on Mutation Spectrum and Therapeutic Strategies. Curr. Pharm. Des. 2018, 24, 3566–3575. [Google Scholar] [CrossRef] [PubMed]
- McGowan, E.M.; Lin, Y.; Hatoum, D. Good Guy or Bad Guy? The Duality of Wild-Type p53 in Hormone-Dependent Breast Cancer Origin, Treatment, and Recurrence. Cancers 2018, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, N.W.; Yang, L.; Rogan, E.G.; Cavalieri, E.L. Evidence for NQO2-mediated reduction of the carcinogenic estrogen ortho-quinones. Free Radic. Biol. Med. 2009, 46, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, M.; Vaclavikova, R.; Ehrlichova, M.; Mrhalova, M.; Kodet, R.; Kubackova, K.; Vrána, D.; Gut, I.; Soucek, P. Association of superoxide dismutases and NAD(P)H quinone oxidoreductases with prognosis of patients with breast carcinomas. Int. J. Cancer 2012, 130, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.-D.; Di, G.-H.; Fan, L.; Hu, Z.; Chen, A.-X.; Shao, Z.-M. Caution regarding genotyping methodology for a tri-allelic polymorphism in the novel breast cancer susceptibility gene NQO2. Breast Cancer Res. Treat. 2009, 118, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-Q.; Zhu, S.-Y.; He, Y.; Yu, K.-D. Association Between a Tri-allelic Polymorphism in the Estrogen Metabolism Oxidoreductase NRH:Quinone Oxidoreductase 2 Gene and Risk of Breast Cancer by Molecular Subtype. Front. Genet. 2021, 12, 658285. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, P.; Ventura, M.; Baudi, F.; Cucinotto, I.; Arbitrio, M.; Di Martino, M.T.; Tassone, P. BRCA1/2 genetic background-based therapeutic tailoring of human ovarian cancer: Hope or reality? J. Ovarian Res. 2009, 2, 14. [Google Scholar] [CrossRef]
- Doonan, B.B.; Schaafsma, E.; Pinto, J.T.; Wu, J.M.; Hsieh, T.-C. Application of open-access databases to determine functional connectivity between resveratrol-binding protein QR2 and colorectal carcinoma. In Vitro Cell. Dev. Biol. Anim. 2017, 53, 575–578. [Google Scholar] [CrossRef]
- Sanchez Calle, A.; Kawamura, Y.; Yamamoto, Y.; Takeshita, F.; Ochiya, T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018, 109, 2093–2100. [Google Scholar] [CrossRef]
- Chen, D.; Sun, Q.; Cheng, X.; Zhang, L.; Song, W.; Zhou, D.; Lin, J.; Wang, W. Genome-wide analysis of long noncoding RNA (lncRNA) expression in colorectal cancer tissues from patients with liver metastasis. Cancer Med. 2016, 5, 1629–1639. [Google Scholar] [CrossRef]
- Hsieh, T.-C. Antiproliferative effects of resveratrol and the mediating role of resveratrol targeting protein NQO2 in androgen receptor-positive, hormone-non-responsive CWR22Rv1 cells. Anticancer Res. 2009, 29, 3011–3017. [Google Scholar] [PubMed]
- Hsieh, T.-C.; Lin, C.-Y.; Bennett, D.J.; Wu, E.; Wu, J.M. Biochemical and cellular evidence demonstrating AKT-1 as a binding partner for resveratrol targeting protein NQO2. PLoS ONE 2014, 9, e101070. [Google Scholar] [CrossRef] [PubMed]
- Dorai, T.; Shah, A.; Summers, F.; Mathew, R.; Huang, J.; Hsieh, T.-C.; Wu, J.M. NRH:quinone oxidoreductase 2 (NQO2) and glutaminase (GLS) both play a role in large extracellular vesicles (LEV) formation in preclinical LNCaP-C4-2B prostate cancer model of progressive metastasis. Prostate 2018, 78, 1181–1195. [Google Scholar] [CrossRef] [PubMed]
- Renaud, C.O.; Ziros, P.G.; Chartoumpekis, D.V.; Bongiovanni, M.; Sykiotis, G.P. Keap1/Nrf2 Signaling: A New Player in Thyroid Pathophysiology and Thyroid Cancer. Front. Endocrinol. 2019, 10, 510. [Google Scholar] [CrossRef]
- Iida, A.; Sekine, A.; Saito, S.; Kitamura, Y.; Kitamoto, T.; Osawa, S.; Mishima, C.; Nakamura, Y. Catalog of 320 single nucleotide polymorphisms (SNPs) in 20 quinone oxidoreductase and sulfotransferase genes. J. Hum. Genet. 2001, 46, 225–240. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Inyushin, M.Y.; Huertas, A.; Kucheryavykh, Y.V.; Kucheryavykh, L.Y.; Tsydzik, V.; Sanabria, P.; Eaton, M.J.; Skatchkov, S.N.; Rojas, L.V.; Wessinger, W.D. L-DOPA Uptake in Astrocytic Endfeet Enwrapping Blood Vessels in Rat Brain. Park. Dis. 2012, 2012, 321406. [Google Scholar] [CrossRef]
- Lieu, C.A.; Chinta, S.J.; Rane, A.; Andersen, J.K. Age-related behavioral phenotype of an astrocytic monoamine oxidase-B transgenic mouse model of Parkinson’s disease. PLoS ONE 2013, 8, e54200. [Google Scholar] [CrossRef]
- Benoit, C.-E.; Bastianetto, S.; Brouillette, J.; Tse, Y.; Boutin, J.A.; Delagrange, P.; Wong, T.; Sarret, P.; Quirion, R. Loss of quinone reductase 2 function selectively facilitates learning behaviors. J. Neurosci. 2010, 30, 12690–12700. [Google Scholar] [CrossRef]
- Hashimoto, T.; Nakai, M. Increased hippocampal quinone reductase 2 in Alzheimer’s disease. Neurosci. Lett. 2011, 502, 10–12. [Google Scholar] [CrossRef]
- Gould, N.L.; Scherer, G.R.; Carvalho, S.; Shurrush, K.; Kayyal, H.; Edry, E.; Elkobi, A.; David, O.; Foqara, M.; Thakar, D.; et al. Specific quinone reductase 2 inhibitors reduce metabolic burden and reverse Alzheimer’s disease phenotype in mice. J. Clin. Investig. 2023, 133, e162120. [Google Scholar] [CrossRef] [PubMed]
- Gould, N.L.; Elkobi, A.; Edry, E.; Daume, J.; Rosenblum, K. Muscarinic-Dependent miR-182 and QR2 Expression Regulation in the Anterior Insula Enables Novel Taste Learning. eNeuro 2020, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [PubMed]
- Brouillette, J.; Young, D.; During, M.J.; Quirion, R. Hippocampal gene expression profiling reveals the possible involvement of Homer1 and GABAB receptors in scopolamine-induced amnesia. J. Neurochem. 2007, 102, 1978–1989. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, A.N.; Jacob, E.; Sharma, V.; Inberg, S.; Elkobi, A.; Ounallah-Saad, H.; Pasmanik-Chor, M.; Edry, E.; Rosenblum, K. Expression of Quinone Reductase-2 in the Cortex Is a Muscarinic Acetylcholine Receptor-Dependent Memory Consolidation Constraint. J. Neurosci. 2015, 35, 15568–15581. [Google Scholar] [CrossRef] [PubMed]
- Arbitrio, M.; Scionti, F.; Di Martino, M.T.; Pensabene, L.; Tassone, P.; Tagliaferri, P. Pharmacogenetics/Pharmacogenomics of Drug-Metabolizing Enzymes and Transporters. In Comprehensive Pharmacology; Kenakin, T., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 657–697. [Google Scholar] [CrossRef]
- Principi, N.; Petropulacos, K.; Esposito, S. Impact of Pharmacogenomics in Clinical Practice. Pharmaceuticals 2023, 16, 1596. [Google Scholar] [CrossRef]
- Arbitrio, M.; Scionti, F.; Di Martino, M.T.; Caracciolo, D.; Pensabene, L.; Tassone, P.; Tagliaferri, P. Pharmacogenomics Biomarker Discovery and Validation for Translation in Clinical Practice. Clin. Transl. Sci. 2021, 14, 113–119. [Google Scholar] [CrossRef]
- de Jong, L.M.; Jiskoot, W.; Swen, J.J.; Manson, M.L. Distinct Effects of Inflammation on Cytochrome P450 Regulation and Drug Metabolism: Lessons from Experimental Models and a Potential Role for Pharmacogenetics. Genes 2020, 11, 1509. [Google Scholar] [CrossRef]
- Traver, R.D.; Siegel, D.; Beall, H.D.; Phillips, R.M.; Gibson, N.W.; Franklin, W.A.; Ross, D. Characterization of a polymorphism in NAD(P)H: Quinone oxidoreductase (DT-diaphorase). Br. J. Cancer 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Jamieson, D.; Lee, J.; Cresti, N.; Jackson, R.; Griffin, M.; Sludden, J.; Verrill, M.; Boddy, A.V. Pharmacogenetics of adjuvant breast cancer treatment with cyclophosphamide, epirubicin and 5-fluorouracil. Cancer Chemother. Pharmacol. 2014, 74, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Legge, S.E.; Walters, J.T. Genetics of clozapine-associated neutropenia: Recent advances, challenges and future perspective. Pharmacogenomics 2019, 20, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Tenen, D.G.; Hromas, R.; Licht, J.D.; Zhang, D.E. Transcription factors, normal myeloid development, and leukemia. Blood 1997, 90, 489–519. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Eberhard, D.; Abraham, Y.; Bastuck, S.; Boesche, M.; Hobson, S.; Mathieson, T.; Perrin, J.; Raida, M.; Rau, C.; et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044. [Google Scholar] [CrossRef]
- Groß, C.J.; Mishra, R.; Schneider, K.S.; Médard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.-A.; Fromm, S.; et al. K+ Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, Z.; Hou, W.; Jiang, Y.; Liu, G.; Ren, X.; Liu, K.; Liu, H.; Chen, K.; Huang, H. Quantitative Proteomics Explore the Potential Targets and Action Mechanisms of Hydroxychloroquine. Molecules 2022, 27, 5175. [Google Scholar] [CrossRef] [PubMed]
- Alnabulsi, S.; Santina, E.; Russo, I.; Hussein, B.; Kadirvel, M.; Chadwick, A.; Bichenkova, E.V.; Bryce, R.A.; Nolan, K.; Demonacos, C.; et al. Non-symmetrical furan-amidines as novel leads for the treatment of cancer and malaria. Eur. J. Med. Chem. 2016, 111, 33–45. [Google Scholar] [CrossRef]
- Hussein, B.; Ikhmais, B.; Kadirvel, M.; Magwaza, R.N.; Halbert, G.; Bryce, R.A.; Stratford, I.J.; Freeman, S. Discovery of potent 4-aminoquinoline hydrazone inhibitors of NRH:quinoneoxidoreductase-2 (NQO2). Eur. J. Med. Chem. 2019, 182, 111649. [Google Scholar] [CrossRef]
- Lozinskaya, N.A.; Bezsonova, E.N.; Dubar, M.; Melekhina, D.D.; Bazanov, D.R.; Bunev, A.S.; Grigor’eva, O.B.; Klochkov, V.G.; Sokolova, E.V.; Babkov, D.A.; et al. 3-Arylidene-2-oxindoles as Potent NRH:Quinone Oxidoreductase 2 Inhibitors. Molecules 2023, 28, 1174. [Google Scholar] [CrossRef]
- Rashid, M.; Maqbool, A.; Shafiq, N.; Bin Jardan, Y.A.; Parveen, S.; Bourhia, M.; Nafidi, H.-A.; Khan, R.A. The combination of multi-approach studies to explore the potential therapeutic mechanisms of imidazole derivatives as an MCF-7 inhibitor in therapeutic strategies. Front. Chem. 2023, 11, 1197665. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Y.; Li, N.; Liu, W.T.; Liang, J.Z.; Sun, Y.; Zhang, W.X.; Fang, R.D.; Huang, S.L.; Sun, Z.H.; et al. Curcumol Overcomes TRAIL Resistance of Non-Small Cell Lung Cancer by Targeting NRH:Quinone Oxidoreductase 2 (NQO2). Adv. Sci. 2020, 7, 2002306. [Google Scholar] [CrossRef] [PubMed]
- Cura, Y.; Perez Ramirez, C.; Sanchez Martin, A.; Martinez Martinez, F.; Calleja Hernandez, M.A.; Ramirez Tortosa, M.D.C.; Jimenez Morales, A. Genetic polymorphisms on the effectiveness or safety of breast cancer treatment: Clinical relevance and future perspectives. Mutat. Res./Rev. Mutat. Res. 2021, 788, 108391. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L. Phenotypic Modulation of Cancer-Associated Antioxidant NQO1 Activity by Post-Translational Modifications and the Natural Diversity of the Human Genome. Antioxidants 2023, 12, 379. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Xiao, P.; Zhou, B.; Chen, Y.; Kang, L.; Wang, Q.; Lin, J.; Son, M.; Wu, Q. Influence of NQO1 Polymorphisms on Warfarin Maintenance Dose: A Systematic Review and Meta-Analysis (rs1800566 and rs10517). Cardiovasc. Ther. 2021, 2021, 5534946. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janda, E.; Boutin, J.A.; De Lorenzo, C.; Arbitrio, M. Polymorphisms and Pharmacogenomics of NQO2: The Past and the Future. Genes 2024, 15, 87. https://doi.org/10.3390/genes15010087
Janda E, Boutin JA, De Lorenzo C, Arbitrio M. Polymorphisms and Pharmacogenomics of NQO2: The Past and the Future. Genes. 2024; 15(1):87. https://doi.org/10.3390/genes15010087
Chicago/Turabian StyleJanda, Elzbieta, Jean A. Boutin, Carlo De Lorenzo, and Mariamena Arbitrio. 2024. "Polymorphisms and Pharmacogenomics of NQO2: The Past and the Future" Genes 15, no. 1: 87. https://doi.org/10.3390/genes15010087
APA StyleJanda, E., Boutin, J. A., De Lorenzo, C., & Arbitrio, M. (2024). Polymorphisms and Pharmacogenomics of NQO2: The Past and the Future. Genes, 15(1), 87. https://doi.org/10.3390/genes15010087