
Utilizing Massively Parallel Sequencing (MPS) of Human Leukocyte Antigen (HLA) Gene Polymorphism to Assess Relatedness in Deficiency Parentage Testing
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. CE-Based aSTR Genotyping
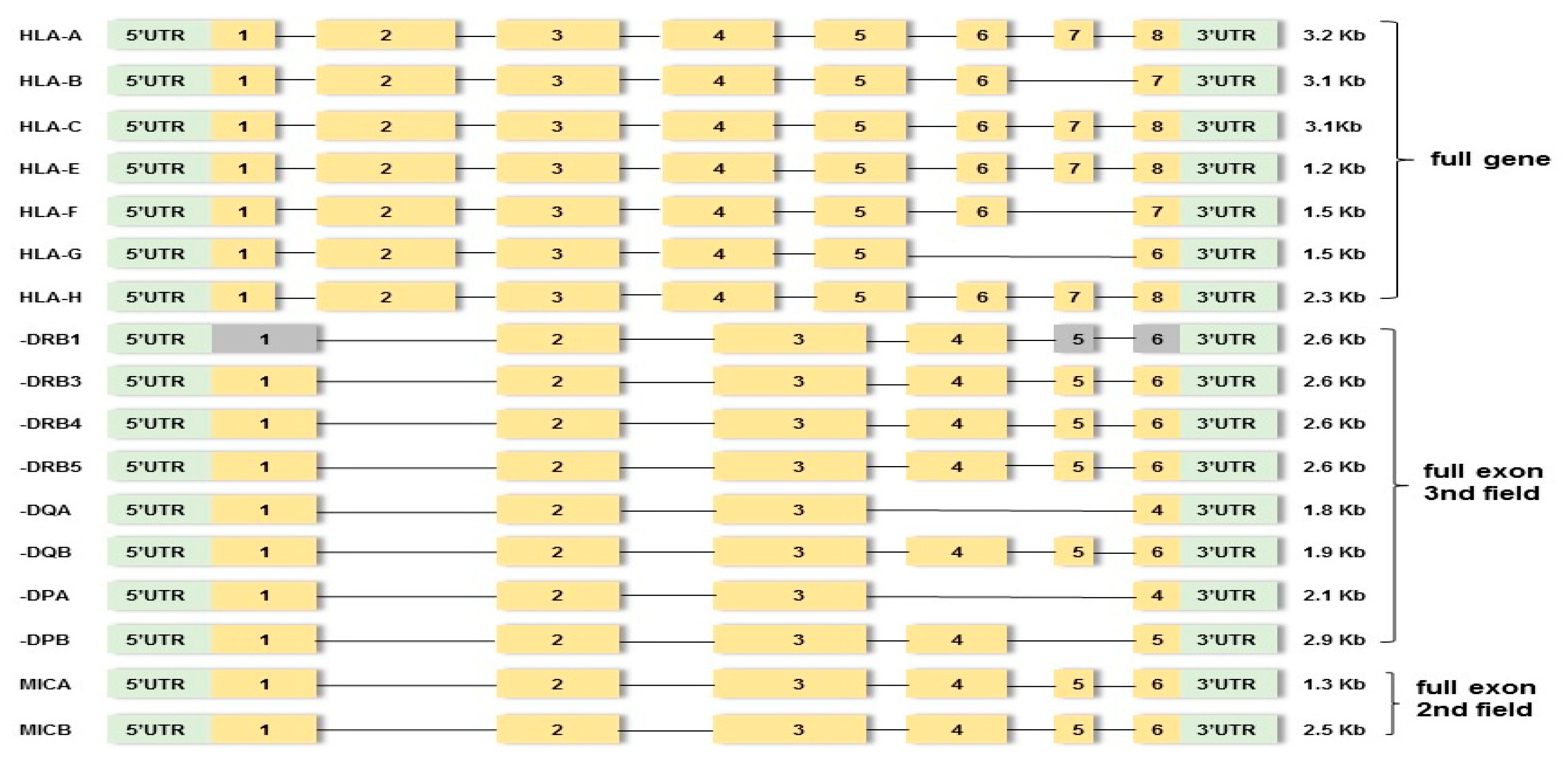
2.3. NGS-Based HLA Genotyping
2.3.1. HLA Gene Amplification and Library Preparation
2.3.2. HLA Sequencing
2.4. Statistical Analysis
2.4.1. Data Analysis for aSTRs Markers
2.4.2. Data Analysis for HLA Alleles
3. Results
3.1. aSTR Typing Results
3.2. HLA Typing Results
3.2.1. HLA Sequencing Metrics
3.2.2. HLA Haplotypes
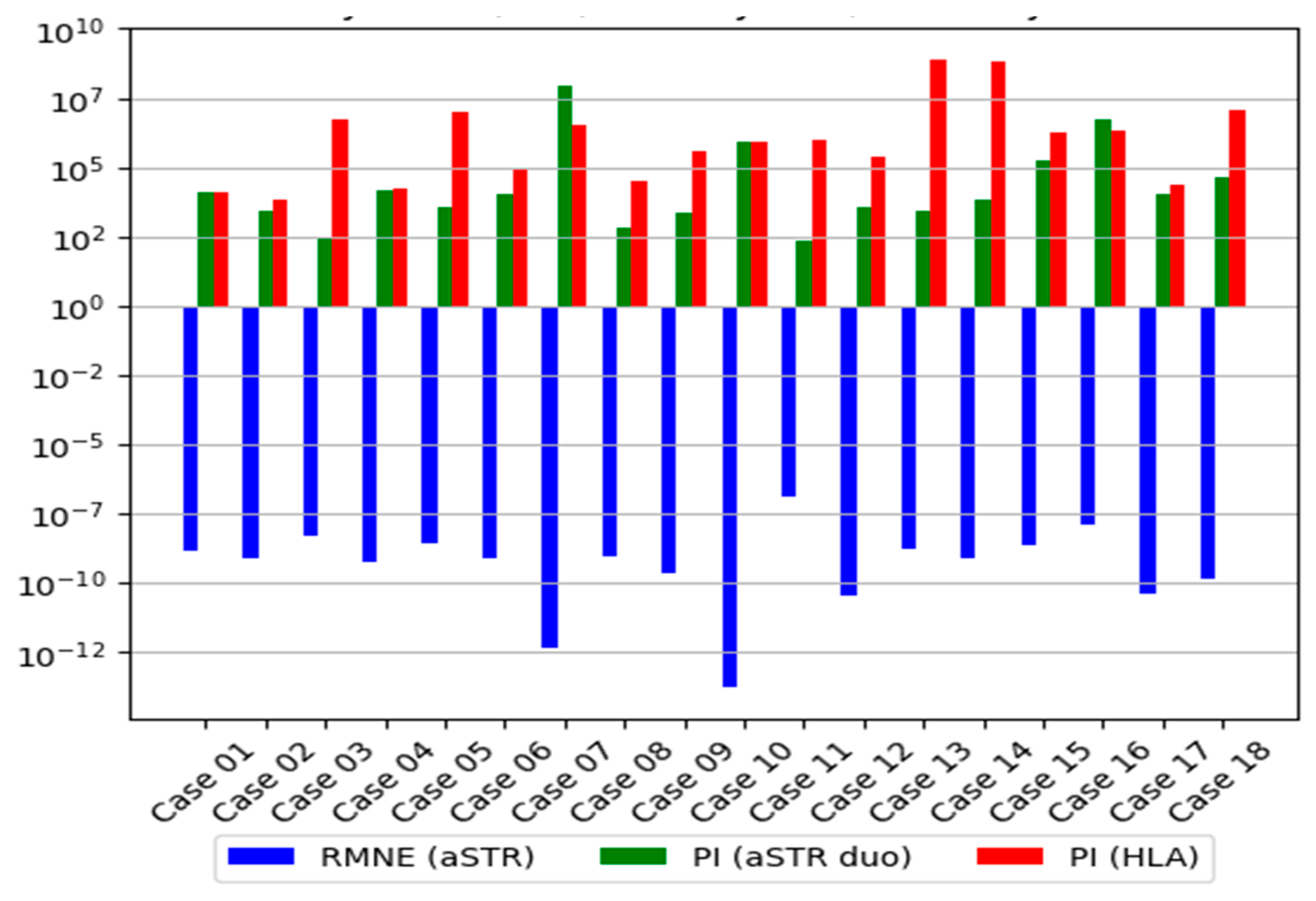
3.2.3. Distribution of the LR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Medhasi, S.; Chantratita, N. Human Leukocyte Antigen (HLA) System: Genetics and Association with Bacterial and Viral Infections. J. Immunol. Res. 2022, 2022, 9710376. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.G.E. Nomenclature for factors of the HLA system, update April, May and June 2023. HLA 2023, 102, 401–430. [Google Scholar] [CrossRef] [PubMed]
- Tumer, G.; Simpson, B.; Roberts, T.K. Genetics, Human Major Histocompatibility Complex (MHC). In StatPearls; Stat Pearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Silver, H. Paternity testing. Crit. Rev. Clin. Lab. Sci. 1989, 27, 391–408. [Google Scholar] [CrossRef]
- Carrington, M. Recombination within the human MHC. Immunol. Rev. 1999, 167, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Kereszturya, L.; Rajczya, K.; Lászikb, A.; Gyódia, E.; Pénzes, M.; Falus, A.; Petrányia, G.G. Combination of DNA-based and conventional methods to detect human leukocyte antigen polymorphism and its use for paternity testing. Am. J. Forensic Med. Pathol. 2002, 23, 57–62. [Google Scholar] [CrossRef]
- Toscanini, U.; Berardi, G.; Rodríguez-Diez, M.B.; Padros, K.; Raimondi, E. Molecular HLA typing is an invaluable tool in some complex paternity cases. Forensic Sci. Int. Genet. Suppl. Ser. 2008, 1, 531–532. [Google Scholar] [CrossRef]
- Grubic, Z.; Stingl, K.; Martinez, N.; Palfi, B.; Brkljacic-Kerhin, V.; Kastelan, A. STR and HLA analysis in paternity testing. Int. Congr. Ser. 2004, 1261, 535–537. [Google Scholar] [CrossRef]
- Ota, M.; Shimada, K.; Asamura, H.; Takayanag, K.; Katsuyama, Y.; Fukushima, H. Validation of sensitive human leukocyte antigen-sequence-specific primer and probe typing in forensic DNA examination. Leg. Med. 2006, 8, 203–209. [Google Scholar] [CrossRef]
- Dean, L.; Kwon, Y.J.; Philpott, M.K.; Stanciu, C.E.; Seashols-Williams, S.J.; Dawson Cruz, T.; Sturgill, J.; Ehrhardt, C.J. Separation of uncompromised whole blood mixtures for single source STR profiling using fluorescently-labeled human leukocyte antigen (HLA) probes and fluorescence activated cell sorting (FACS). Forensic Sci. Int. Genet. 2015, 17, 8–16. [Google Scholar] [CrossRef]
- Pokhriyal, B.; Thorat, K.; Limaye, D.A.; Joshi, Y.M.; Kadam, V.J.; Dubey, R. Microsatellite markers—A novel tool in molecular genetics. Int. J. Res. Pharm. Chem. 2012, 2, 397–412. [Google Scholar]
- Fan, H.; Chu, J.Y. A brief review of short tandem repeat mutation. Genom. Proteom. Bioinform. 2007, 5, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, H.; Wang, Q.K.; Wang, P.; Wang, F.; Xu, C. Loss of heterozygosity detected at three short tandem repeat locus commonly used for human DNA identification in a case of paternity testing. Leg. Med. 2017, 24, 7–11. [Google Scholar] [CrossRef]
- Dauber, E.M.; Kratzer, A.; Neuhuber, F.; Parson, W.; Klintschar, M.; Bär, W.; Mayr, W.R. Germline mutations of STR-alleles include multi-step mutations as defined by sequencing of repeat and flanking regions. Forensic Sci. Int. Genet. 2012, 6, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, A.; Fuentes, F.F. Mutational dynamics of microsatellites. Mol. Biotechnol. 2010, 44, 250–266. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, B.; Klintschar, M.; Neuhuber, F.; Hühne, J.; Rolf, B. Mutation rate in human microsatellites: Influence of the structure and length of the tandem repeat. Am. J. Hum. Genet. 1998, 62, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Kouniaki, D.I.; Papasteriades, C.A.; Tsirogianni, A. Short Tandem Repeats Loci in Parentage Testing. Hosp. Chron. 2015, 10, 83–90. [Google Scholar]
- Chaitanya, L.; van Oven, M.; Brauer, S.; Zimmermann, B.; Huber, G.; Xavier, C.; Parson, W.; de Knijff, P.; Kayser, M. High-quality mtDNA control region sequences from 680 individuals sampled across the Netherlands to establish a national forensic mtDNA reference database. Forensic Sci. Int. Genet. 2016, 21, 158–167. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhou, Z.; Wang, L.; Quan, C.; Liu, Q.; Tang, Z.; Liu, L.; Liu, Y.; Wang, S. Pairwise kinship testing with a combination of STR and SNP loci. Forensic Sci. Int. Genet. 2020, 46, 102265. [Google Scholar] [CrossRef]
- Kaitholia, K.; Dash, H.R.; Shrivastava, P.; Kumawat, R.K.; Dixit, S.; Chaubey, G. Forensic characterization and genetic evaluation in the Central Indian population using 27 Y-STRs. Int. J. Leg. Med. 2021, 135, 791–792. [Google Scholar] [CrossRef]
- Robinson, J.; Barker, D.J.; Georgiou, X.; Cooper, M.A.; Flicek, P.; Marsh, S.G.E. IPD-IMGT/HLA Database. Nucleic Acids Res. 2020, 48, D948–D955. [Google Scholar] [CrossRef]
- Creary, L.E.; Guerra, S.G.; Chong, W.; Brown, C.J.; Turner, T.R.; Robinson, J.; Bultitude, W.P.; Mayor, N.P.; Marsh, S.G.E.; Saito, K.; et al. Next-generation HLA typing of 382 International Histocompatibility Working Group reference B-lymphoblastoid cell lines: Report from the 17th International HLA and Immunogenetics Workshop. Hum. Immunol. 2019, 80, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.N.; Sherwood, K.R.; Mostafa, A.; Benedicto, R.V.; ElaAlim, A.; Greenshields, A.; Keown, P.; Liwski, R.; Lan, J.H. Novel alleles in the era of next-generation sequencing-based HLA typing calls for standardization and policy. Front. Genet. 2023, 14, 1282834. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Xu, F.; Wu, J.; Schubert, J.; Li, M.M. Application of Next Generation Sequencing in Laboratory Medicine. Ann. Lab. Med. 2021, 41, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Gjertson, D.W.; Brenner, C.H.; Baur, M.P.; Carracedo, A.; Guidet, F.; Luque, J.A.; Lessig, R.; Mayr, W.R.; Pascali, V.L.; Prinz, M.; et al. ISFG: Recommendations on biostatistics in paternity testing. Forensic Sci. Int. Genet. 2007, 1, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Scarpetta, M.A.; Staub, R.W.; Einum, D.D. Assessing exclusionary power of a paternity test involving a pair of alleged grandparents. Transfusion 2007, 47, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, M.; Giannuzzi, M.R.; Panarello, D. DNA test to assess criminal responsibility: A Bayesian approach. Qual. Quant. 2018, 52, 2837–2853. [Google Scholar] [CrossRef]
- Steffen, C.R.; Coble, M.D.; Gettings, K.B.; Vallone, P.M. Corrigendum to ‘U.S. Population Data for 29 Autosomal STR Loci’. Forensic Sci. Int. Genet. 2013, 7, e82–e83, Erratum in Forensic Sci. Int. Genet. 2017, 31, e36–e40. [Google Scholar] [CrossRef]
- References AABB-American Association of Blood Banks. Annual Report Summary for Testing in 2008: Relationship Testing Program Unit. pp. 1–64. Available online: https://www.aabb.org/docs/default-source/default-document-library/accreditation/rtannrpt08.pdf?sfvrsn=982c3264_0 (accessed on 1 January 2010).
- Vicard, P.; Dawid, A.P.; Mortera, J.; Lauritzen, S.L. Estimating mutation rates from paternity casework. Forensic Sci. Int. Genet. 2008, 2, 9–18. [Google Scholar] [CrossRef]
- Ge, J.; Eisenberg, A.; Budowle, B. Developing criteria and data to determine best options for expanding the core CODIS loci. Investig. Genet. 2012, 3, 1. [Google Scholar] [CrossRef]
- Egeland, T.; Mostad, P.F. Statistical Genetics and Genetical Statistics: A Forensic Perspective. Scand. J. Stat. 2002, 29, 297–307. [Google Scholar] [CrossRef]
- García-Aceves, M.E.; Romero Rentería, O.; Díaz-Navarro, X.X.; Rangel-Villalobos, H. Paternity tests in Mexico: Results obtained in 3005 cases. J. Forensic Leg. Med. 2018, 55, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Su, Q.; Luo, H.; Li, Y.; Wu, J.; Yan, J.; Hou, Y.; Liang, W.; Zhang, L. Mutational analysis of 33 autosomal short tandem repeat (STR) loci in southwest Chinese Han population based on trio parentage testing. Forensic Sci. Int. Genet. 2016, 23, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.G.E.; Albert, E.D.; Bodmer, W.F.; Bontrop, R.E.; Dupont, B.; Erlich, H.A.; Fernández-Viña, M.; Geraghty, D.E.; Holdsworth, R.; Hurley, C.K.; et al. Nomenclature for Factors of the HLA System, 2010. Tissue Antigens 2010, 75, 291–455. [Google Scholar] [CrossRef] [PubMed]
- González-Andrade, F.; Sánchez, D.; Penacino, G.; Martínez Jarreta, B. Two fathers for the same child: A deficient paternity case of false inclusion with autosomic STRs. Forensic Sci. Int. Genet. 2009, 3, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Narkuti, V.; Vellanki, R.N.; Anubrolu, N.; Doddapaneni, K.K.; Gandhi Kaza, P.C.; Mangamoori, L.N. Single and double incompatibility at vWA and D8S1179/D21S11 loci between mother and child: Implications in kinship analysis. Clin. Chim. Acta 2008, 395, 162–165. [Google Scholar] [CrossRef]
- De Kock, A.; Kloppers, J.J. The impact of motherless paternity testing in a South African population. Afr. Health Sci. 2021, 21, 379–384. [Google Scholar] [CrossRef]
- Aguiar, V.R.C.; de Castro, A.M.; Pinto, L.M.; Ferreira, A.C.S.; Dos Santos, E.V.W.; Louro, I.D. Assessing false paternity risk in simulated motherless cases from more than 20 000 real exclusion trios. Transfusion 2021, 61, 678–681. [Google Scholar] [CrossRef]
- Li, R.; Li, H.; Peng, D.; Hao, B.; Wang, Z.; Huang, E.; Wu, R.; Sun, H. Improved pairwise kinship analysis using massively parallel sequencing. Forensic Sci. Int. Genet. 2019, 38, 77–85. [Google Scholar] [CrossRef]
- Butler, J. Recent advances in forensic biology and forensic DNA typing: INTERPOL review 2019–2022. Forensic Sci. Int. Synerg. 2022, 27, 100311. [Google Scholar] [CrossRef]
- Wu, R.; Li, H.; Peng, D.; Li, R.; Zhang, Y.; Hao, B.; Huang, E.; Zheng, C.; Sun, H. Revisiting the potential power of human leukocyte antigen (HLA) genes on relationship testing by massively parallel sequencing-based HLA typing in an extended family. J. Hum. Genet. 2019, 64, 29–38. [Google Scholar] [CrossRef]
- Duke, J.L.; Mosbruger, T.L.; Ferriola, D.; Chitnis, N.; Hu, T.; Tairis, N.; Margolis, D.J.; Monos, D.S. Resolving MiSeq-Generated Ambiguities in HLA-DPB1 Typing by Using the Oxford Nanopore Technology. J. Mol. Diagn. 2019, 21, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Osoegawa, K.; Vayntrub, T.A.; Wenda, S.; De Santis, D.; Barsakis, K.; Ivanova, M.; Hsu, S.; Barone, J.; Holdsworth, R.; Diviney, M.; et al. Quality control project of NGS HLA genotyping for the 17th International HLA and Immunogenetics Workshop. Hum. Immunol. 2019, 80, 228–236. [Google Scholar] [CrossRef]
- Ingram, K.J.; O’Shields, E.F.; Kiger, D.F.; Gautreaux, M.D. NGS and HLA: The long road ahead. Hum. Immunol. 2020, 81, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Kouniaki, D.; Tarassi, K.; Kitsiou, V.; Fotopoulos, K.; Tsirogianni, A. Identification of the novel HLA-DPB1*02:01:68 allele in a Greek individual. HLA 2023, 102, 550–552. [Google Scholar] [CrossRef] [PubMed]
- Kouniaki, D.; Athanassiades, T.; Fotopoulos, K.; Tsirogianni, A. Genomic sequence of the HLA-A*24:587 allele identified in a Greek individual. HLA 2023, 102, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Paech, C.; Albrecht, V.; Putke, K.; Schöfl, G.; Schöne, B.; Schmidt, A.H.; Lange, V.; Klussmeier, A. HLA-E diversity unfolded: Identification and characterization of 170 novel HLA-E alleles. HLA 2021, 97, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Ehrenberg, P.K.; Geretz, A.; Sindhu, R.K.; Vayntrub, T.; Fernández Viña, M.A.; Apps, R.; Michael, N.L.; Thomas, R. High-throughput next-generation sequencing to genotype six classical HLA loci from 96 donors in a single MiSeq run. HLA 2017, 90, 284–291. [Google Scholar] [CrossRef]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Bravo-Egana, V.; Monos, D. The impact of next-generation sequencing in immunogenetics: Current status and future directions. Curr. Opin. Organ Transpl. 2017, 22, 400–406. [Google Scholar] [CrossRef]
- Kouniaki, D.; Tsirogianni, A. Classical and Non-Classical HLA Alleles as Supplementary Markers in Indirect Kinship Parentage Testing. Immuno 2022, 2, 501–505. [Google Scholar] [CrossRef]
- Kuffel, A.; Gray, A.; Nic Daeid, N. Human Leukocyte Antigen alleles as an aid to STR in complex forensic DNA samples. Sci. Justice 2020, 60, 1–8. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| aSTRs Loci | Alleged Father Genotype | Mother Genotype | Child Genotype | POA | Putative Mutation | Gain/Loss | Sex/Age (Years) | Single/Double Step | |
|---|---|---|---|---|---|---|---|---|---|
| Case 01 | SE33 | 18, 20 | 20, 34 | 19, 34 | 18 or 20 | 18→19 οr 20→19 | Gain or loss | Male/45 | Single |
| Case 02 | SE33 | 15, 34 | 12, 16 | 16, 16 | 15 | 15→16 | Gain | Male/29 | Single |
| Case 03 | SE33 | 17, 25.2 | 19, 28.2 | 19, 24.2 | 25.2 | 25.2→24.2 | Loss | Male/39 | Single |
| Case 04 | SE33 | 28.2, 33.2 | 22.2, 28.2 | 28.2, 32.2 | 33.2 | 33.2→32.2 | Loss | Male/41 | Single |
| Case 05 | SE33 | 20, 20 | 16, 32.2 | 19, 32.2 | 20 | 20→19 | Loss | Male/39 | Single |
| Case 06 | vWA | 17, 19 | 16, 18 | 18, 20 | 19 | 19→20 | Gain | Male/41 | Single |
| Case 07 | vWA | 16, 19 | 16, 17 | 17, 18 | 19 | 19→18 | Loss | Male/36 | Single |
| Case 08 | vWA | 17, 20 | NA | 18, 19 | Uncertain | Uncertain | Gain or loss | Male/33 | Single |
| Case 09 | D12S391 | 16, 21 | 22, 22 | 20, 22 | 21 | 21→20 | Loss | Male/31 | Single |
| Case 10 | D12S391 | 21, 24 | 20, 20 | 20, 20 | 21 | 21→20 | Loss | Male/35 | Single |
| Case 11 | D12S391 | 22, 24 | 19.3, 22 | 19.3, 23 | 22 or 24 | 22→23 οr 24→23 | Gain or loss | Male/28 | Single |
| Case 12 | D8S1179 | 13, 13 | 12, 12 | 12, 14 | 13 | 13→14 | Gain | Male/49 | Single |
| Case 13 | D8S1179 | 13, 15 | 8, 13 | 8, 16 | 15 | 15→16 | Gain | Male/19 | Single |
| Case 14 | D10S1248 | 14, 17 | NA | 15, 16 | Uncertain | Uncertain | Gain or loss | Male/45 | Single |
| Case 15 | D3S1358 | 15, 18 | 18, 18 | 18, 19 | 18 | 18→19 | Gain | Male/41 | Single |
| Case 16 | FGA | 19, 23 | 20, 23 | 22, 23 | 23 | 23→22 | Loss | Male/35 | Single |
| Case 17 | D2S1338 | 16, 24 | 24, 25 | 23, 25 | 24 | 24→23 | Loss | Male/40 | Single |
| Case 18 | D19S433 | 15, 15 | 13, 14.2 | 13, 13 | 15 | 15→13 | Loss | Male/27 | Double |
| aSTR Trio | aSTR Duo * | HLA | aSTR Duo and HLA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RMNE | PE | PI | W | PI | W | PI | W | PI | W | |
| Case 01 | 1.39 × 10−9 | 0.99999999 | 6.66 × 105 | 0.99999849 | 11,732 | 0.99991477 | 13,355 | 0.99992513 | 1.57 × 108 | 0.99999999 |
| Case 02 | 7.82 × 10−10 | 0.99999999 | 1.22 × 105 | 0.99999179 | 2540 | 0.99960645 | 6672 | 0.99985014 | 1.69 × 107 | 0.99999994 |
| Case 03 | 4.90 × 10−9 | 0.99999999 | 5.91 × 105 | 0.99999830 | 260 | 0.99616858 | 5.04 × 106 | 0.99999980 | 1.31 × 109 | 0.99999999 |
| Case 04 | 5.54 × 10−10 | 0.99999999 | 3.78 × 106 | 0.99999973 | 13,925 | 0.99992819 | 16,193 | 0.99993825 | 2.25 × 108 | 0.99999999 |
| Case 05 | 2.85 × 10−9 | 0.99999999 | 1.72 × 105 | 0.99999418 | 3769 | 0.99973475 | 9.63 × 106 | 0.99999989 | 3.63 × 1010 | 0.99999999 |
| Case 06 | 8.17 × 10−10 | 0.99999999 | 2.43 × 107 | 0.99999995 | 10,798 | 0.99990740 | 81,867 | 0.99998778 | 8.83 × 108 | 0.99999999 |
| Case 07 | 4.81 × 10−13 | 0.99999999 | 3.98 × 109 | 0.99999999 | 9.25 × 107 | 0.99999999 | 3.48 × 106 | 0.99999971 | 3.21 × 1014 | 0.99999999 |
| Case 08 | 9.68 × 10−10 | 0.99999999 | NA | NA | 697 | 0.99856734 | 30,769 | 0.99996750 | 2.14 × 107 | 0.99999995 |
| Case 09 | 2.14 × 10−10 | 0.99999999 | 1.29 × 105 | 0.99999224 | 2402 | 0.99958385 | 4.07 × 105 | 0.99999754 | 9.77 × 108 | 0.99999999 |
| Case 10 | 1.82 × 10−14 | 0.99999999 | 4.79 × 107 | 0.99999998 | 8.35 × 105 | 0.99999880 | 8.72 × 105 | 0.99999885 | 7.28 × 1011 | 0.99999999 |
| Case 11 | 1.29 × 10−7 | 0.99999978 | 8473 | 0.99988199 | 210 | 0.99526066 | 1.03 × 106 | 0.99999903 | 2.16 × 108 | 0.99999999 |
| Case 12 | 3.64 × 10−11 | 0.99999999 | 5.44 × 106 | 0.99999982 | 3631 | 0.99972467 | 2.53 × 105 | 0.99999605 | 9.19 × 108 | 0.99999999 |
| Case 13 | 1.57 × 10−9 | 0.99999999 | 4.88 × 105 | 0.99999795 | 2864 | 0.99965096 | 8.06 × 108 | 0.99999999 | 2.31 × 1012 | 0.99999999 |
| Case 14 | 7.38 × 10−10 | 0.99999999 | NA | NA | 7035 | 0.99985787 | 6.34 × 108 | 0.99999999 | 4.46 × 1012 | 0.99999999 |
| Case 15 | 2.44 × 10−9 | 0.99999999 | 51,493 | 0.99998058 | 1.64 × 105 | 0.99999391 | 1.72 × 106 | 0.99999941 | 2.83 × 1011 | 0.99999999 |
| Case 16 | 1.23 × 10−8 | 0.99999998 | 85,436 | 0.99998829 | 5.25 × 106 | 0.99999981 | 2.19 × 106 | 0.99999954 | 1.15 × 1013 | 0.99999999 |
| Case 17 | 4.01 × 10−11 | 0.99999999 | 47,478 | 0.99997894 | 11,035 | 0.99990939 | 21,851 | 0.99995424 | 2.41 × 108 | 0.99999999 |
| Case 18 | 1.45 × 10−10 | 0.99999999 | 6.02 × 107 | 0.99999998 | 40,886 | 0.99997554 | 1.23 × 107 | 0.99999999 | 5.04 × 1012 | 0.99999999 |
| HLA Locus | A | B | C | E | F | G | H | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Alleles | 01:01:01:01 02:01:01 02:01:01:01 02:01:01:05 02:02:01:01 02:11:01:01 03:01:01:01 03:02:01:01 11:01:01 11:01:01:01 23:01:01:01 24:02:01:01 24:02:01:04 26:01:01:01 29:02:01:01 30:01:01:01 30:04:01:01 31:01:02:01 32:01:01:01 33:01:01:01 68:01:01:02 68:01:02:02 69:01:01:01 | 07:02:01 07:05:01 13:02:01:01 14:02:01:26 * 15:18:01:02 18:01:01 18:05:01:02 27:05:02:01 35:01:01 35:03:01 35:03:01:01 35:03:01:03 35:08:01 35:08:01:01 35:580 * 37:01:01 38:01:01 39:01:01:05 40:01:02 40:02:01:41 * 40:06:01:13 41:01:01:01 41:02:01:01 44:02:01 44:02:01:01 44:05:01:01 49:01:01:01 49:01:01:04 51:01:01 51:01:01:01 51:01:01:10 51:01:01:11 51:01:01:33 51:07:01 51:08:01:01 52:01:01:01 52:01:01:02 52:01:01:18 55:01:01:01 56:01:01 58:01:01:03 | 01:02:01 01:02:01:01 02:02:02:01 03:02:02 03:02:02:05 03:03:01:01 03:04:01:01 04:01:01:06 04:01:01:14 04:01:01:175 * 04:01:01:75 04:01:01:79 05:01:01 05:01:01:02 06:02:01:01 07:01:01 07:01:01:16 07:02:01 07:02:01:01 07:02:01:03 07:04:01:03 08:02:01:01 12:02:02 12:02:02:01 12:03:01:01 14:02:01:01 15:02:01 15:02:01:01 15:05:02 15:09:01:01 16:02:01:01 16:04:01:01 17:01:01:05 | 01:01:01 01:01:01:06 01:01:01:10 01:03:01:01 01:03:02 01:03:02:01 01:03:03 01:03:05:01 01:06:01:01 | 01:01:01 01:01:01:08 01:01:01:09 01:01:01:18 01:01:02 01:01:02:08 01:01:02:09 01:01:02:10 01:03:01 01:03:01:03 01:04:01:02 | 01:01:01 01:01:01:01 01:01:01:04 01:01:01:05 01:01:02 01:01:02:01 01:01:03:03 01:01:08 01:01:12 01:01:13 01:01:14 01:01:22:01 01:03:01 01:03:01:02 01:04:01:01 01:04:04 01:05N 01:06:01:01 | 01:01:01 01:01:01:01 01:01:02:01 01:03:01:01 02:01:01:01 02:02:01:01 02:03:01 02:03:02 02:04:01 02:05:01:01 02:05:01:03 02:07 02:07:01:01 02:09 02:12 02:23 | |||
| HLA Locus | DPA1 | DPB1 | DQA1 | DQB1 | DRB1 | DRB3 | DRB4 | DRB5 | MICA | MICB |
| Alleles | 01:03:01 01:03:01:60 02:01:01 02:01:01:02 02:01:08 02:07:01 | 01:01:01G 02:01:02 02:01:02G 03:01:01G 04:01:01 04:01:01G 04:02:01 04:02:01G 06:01:01G 10:01:01G 104:01:01 13:01:01 13:01:01G 14:01:01G 23:01:01 | 01:01:01 01:01:02:01 01:02:01 01:02:02 01:03:01 01:03:01:01 01:04:01 01:04:02 01:05:01 02:01:01 02:02:02 03:01:01 03:03:01 05:01:01 05:01:01:03 05:05:01 06:01:01 06:01:01:03 | 02:01:01 02:02:01 02:02:01:02 03:01:01 03:01:01:02 03:01:01G 03:02:01 03:03:02 03:292 05:01:01 05:01:01G 05:02:01 05:03:01 05:03:01G 06:01:01 06:02:01 06:03:01 06:03:01G 06:04:01 06:04:01G | 01:01:01 01:02:01:03 03:01:01 04:01:01 04:02:01 04:05:01 04:07:01 07:01:01 07:07:01:01 08:01:01 08:03:02 08:10 10:01:01 11:01:01 11:03:01 11:04:01 11:04:01G 12:01:01 13:01:01 13:01:01:07 13:02:01 14:01:01G 14:04:01 14:54:01 15:01:01 15:02:01 15:02:02 16:01:01 16:02:01G | 01:01:02 01:01:02G 02:02:01 02:02:01G 03:01:01 | 01:01:01 01:03:01 01:03:01N | 01:01:01 01:02:01 01:10N 02:02:01 | 002:01 004:01 006 007:01 008:01 008:02 008:04 009:01 011:01 012:01 012:03 016:01 018:01 019:01 027:01 049:01 19:01 | 002:01 004:01 005:01 005:02 005:03 008:01 014:01 |
| HLA Novel Allele | Most Homologous Allele | gDNA Position | Exon | Codon Change | NC | SNP | aa Change | Type of Mutation | GenBank Accession Number | IPD-IMGT/HLA Database Accession Number |
|---|---|---|---|---|---|---|---|---|---|---|
| B*14:02:01:26 | B*14:02:01:01 | −260 | 5′UTR | G to T | No | Silent | OQ357851 | HWS10065189 | ||
| B*35:580 | B*35:01:01:02 | 2023 | 5 | 304 (GCT >> GTT) | C to T | Yes Ala (A) to Val (V) | Non-synonymous | OQ885042 | HWS10066175 | |
| B*40:02:01:41 | B*40:02:01:01 | 3010:1 3020 3021 | 3′UTR | C to Deletion (−) C to T C to T | No | Silent | OQ885046 | HWS10066149 | ||
| C*04:01:01:175 | C*04:01:01:01 | −249 | 5′UTR | C to T | No | Silent | OQ885045 | HWS10066153 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouniaki, D.I.; Fotopoulos, K.V.; Tarassi, K.; Tsirogianni, A. Utilizing Massively Parallel Sequencing (MPS) of Human Leukocyte Antigen (HLA) Gene Polymorphism to Assess Relatedness in Deficiency Parentage Testing. Genes 2024, 15, 150. https://doi.org/10.3390/genes15020150
Kouniaki DI, Fotopoulos KV, Tarassi K, Tsirogianni A. Utilizing Massively Parallel Sequencing (MPS) of Human Leukocyte Antigen (HLA) Gene Polymorphism to Assess Relatedness in Deficiency Parentage Testing. Genes. 2024; 15(2):150. https://doi.org/10.3390/genes15020150
Chicago/Turabian StyleKouniaki, Diamanto I., Konstantinos V. Fotopoulos, Katerina Tarassi, and Alexandra Tsirogianni. 2024. "Utilizing Massively Parallel Sequencing (MPS) of Human Leukocyte Antigen (HLA) Gene Polymorphism to Assess Relatedness in Deficiency Parentage Testing" Genes 15, no. 2: 150. https://doi.org/10.3390/genes15020150
APA StyleKouniaki, D. I., Fotopoulos, K. V., Tarassi, K., & Tsirogianni, A. (2024). Utilizing Massively Parallel Sequencing (MPS) of Human Leukocyte Antigen (HLA) Gene Polymorphism to Assess Relatedness in Deficiency Parentage Testing. Genes, 15(2), 150. https://doi.org/10.3390/genes15020150