
Therapeutic Management and Outcomes of Hepatoblastoma in a Pediatric Patient with Mosaic Edwards Syndrome
 , , ,
, , ,
Abstract
:1. Introduction
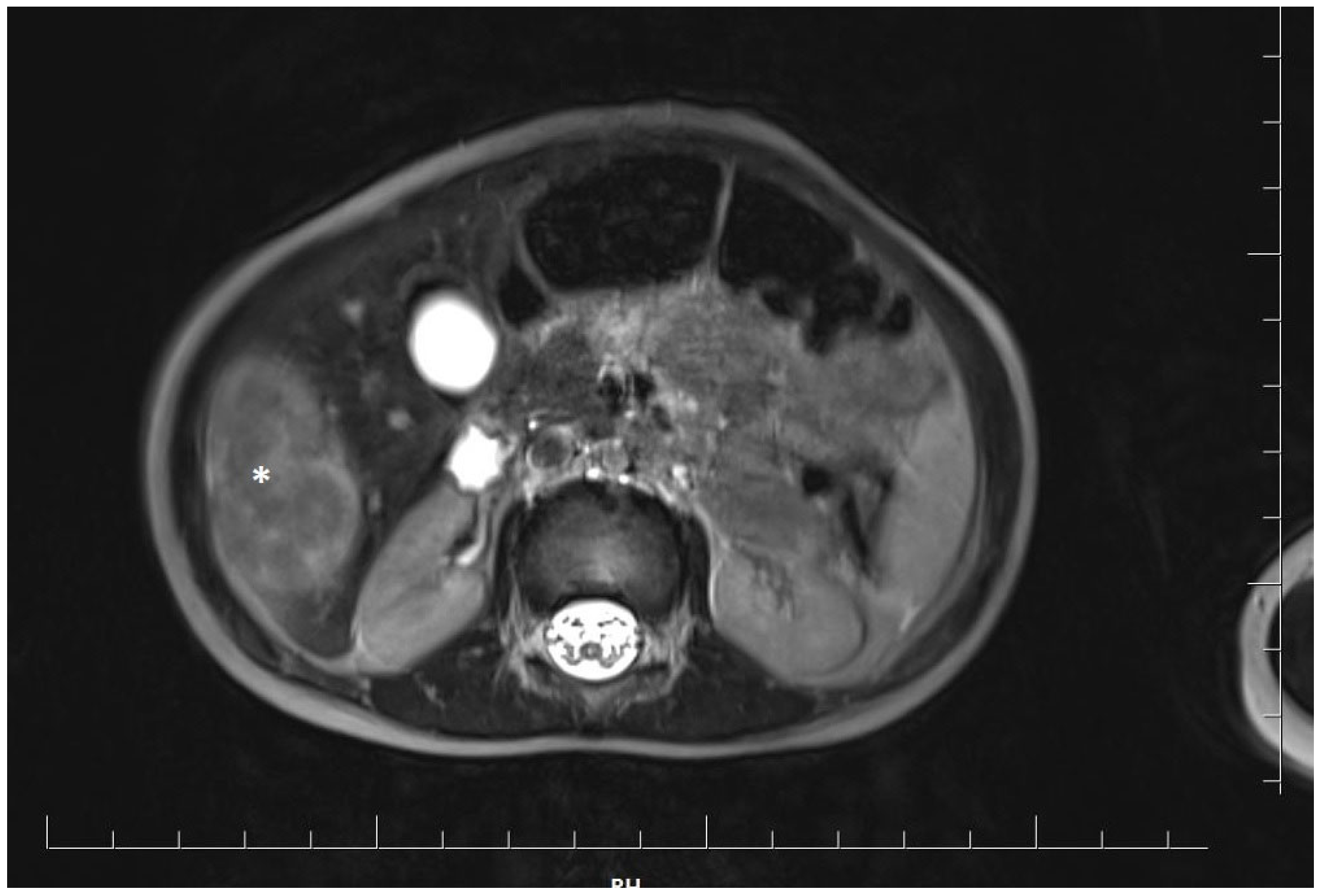
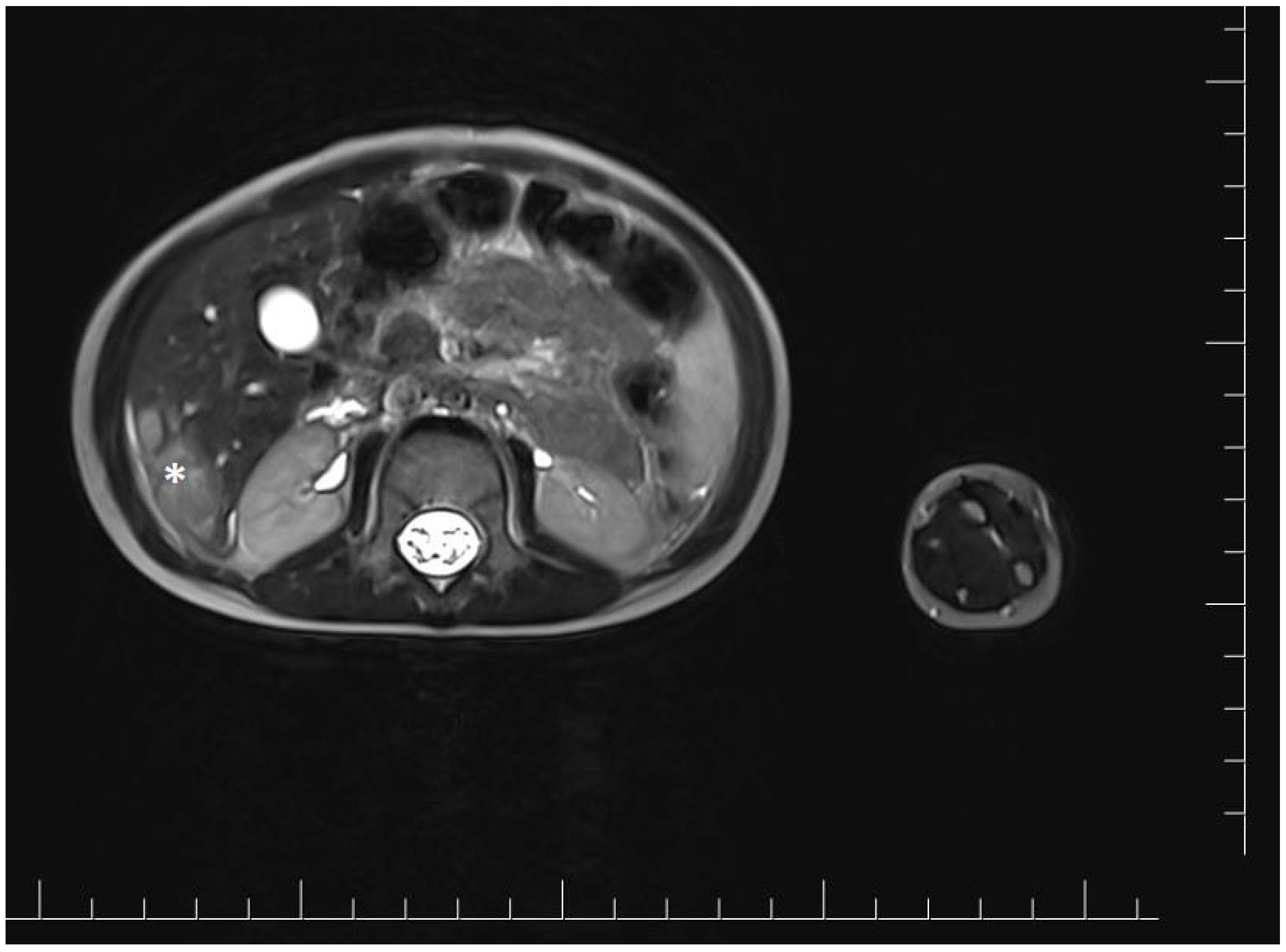
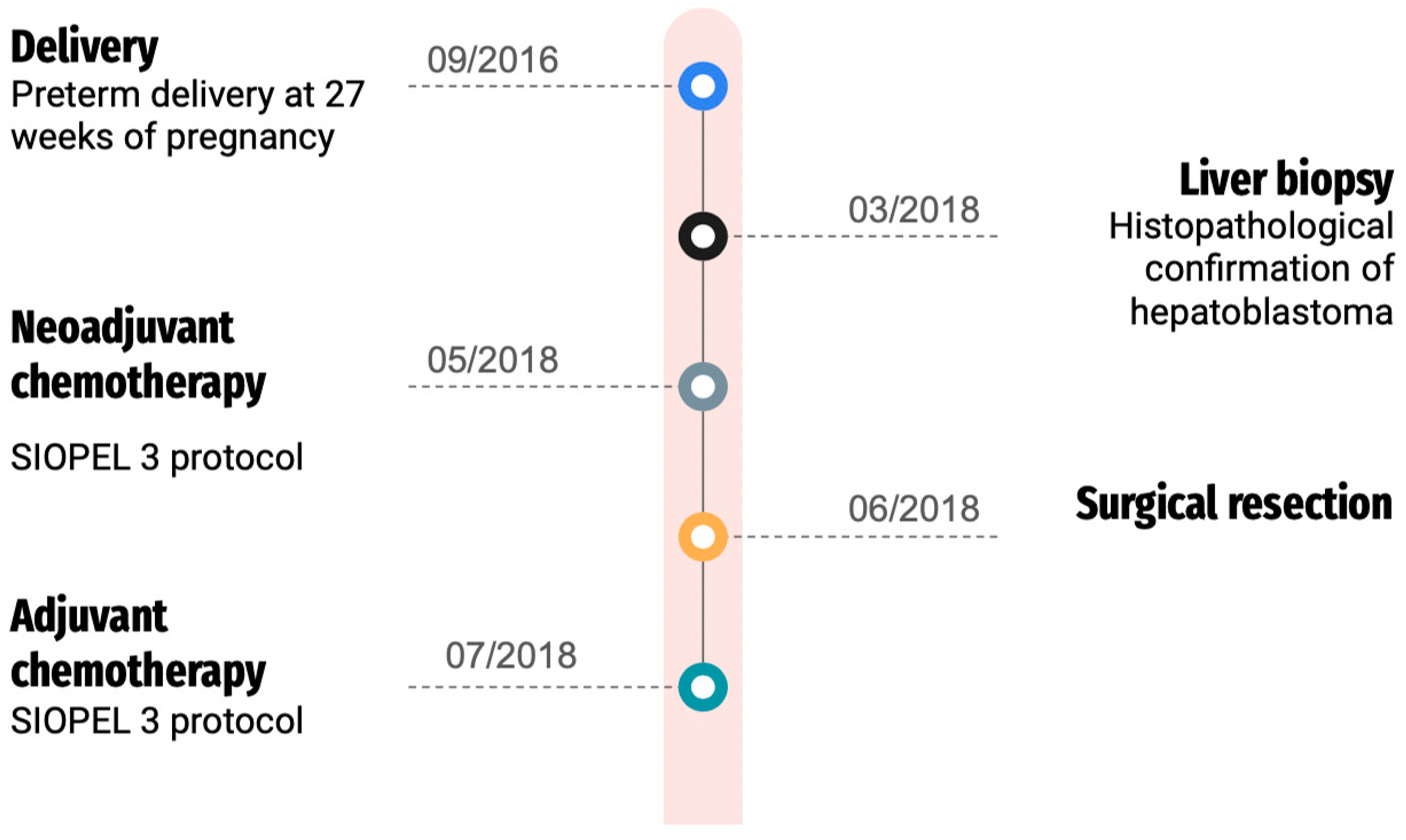
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kepple, J.W.; Fishler, K.P.; Peeples, E.S. Surveillance guidelines for children with trisomy 18. Am. J. Med. Genet. A 2021, 185, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Wheeler, K.; Stewart, H.; Campbell, C. Hepatoblastoma in a mosaic trisomy 18 child with hemihypertrophy. BMJ Case Rep. 2016, 2016, bcr2015211380. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.M.; Marion, R.; Ramesh, K.H.; Kim, J.S.; Ewart, M.; Ricafort, R. Hepatoblastoma in a mosaic trisomy 18 patient. J. Pediatr. Hematol. Oncol. 2012, 34, e145–e148. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.; Dearmun, A. Edwards’ syndrome. Nurs. Child. Young People 2016, 28, 17. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.C. Management of Children with the Trisomy 18 and Trisomy 13 Syndromes: Is there a Shift in the Paradigm of Care? Am. J. Perinatol. 2021, 38, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Outtaleb, F.Z.; Errahli, R.; Imelloul, N.; Jabrane, G.; Serbati, N.; Dehbi, H. La trisomie 18 ou syndrome d’Edwards en post-natal: Étude descriptive au Centre Hospitalier Universitaire de Casablanca et revue de littérature [Trisomy 18 or postnatal Edward´s syndrome: Descriptive study conducted at the University Hospital Center of Casablanca and literature review]. Pan Afr. Med. J. 2020, 37, 309. [Google Scholar] [CrossRef]
- Satgé, D.; Nishi, M.; Sirvent, N.; Vekemans, M. A tumor profile in Edwards syndrome (trisomy 18). Am. J. Med. Genet. C Semin. Med. Genet. 2016, 172, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Kitanovski, L.; Ovcak, Z.; Jazbec, J. Multifocal hepatoblastoma in a 6-month-old girl with trisomy 18: A case report. J. Med. Case Rep. 2009, 3, 8319. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L. (Ed.) Smith’s Recognizable Patterns of Human Malformation, 6th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2009; Volume 13. [Google Scholar]
- Morallo, L.M.; Rosenblum, H.; Esterly, K.L.; Johnson, W.D.; Storlazzi, J.J.; Narvaez, A.C.; Borgaonkar, D.S. Trisomy 18 (Edwards syndrome) in Delaware. Del. Med. J. 1983, 55, 27–30. [Google Scholar] [PubMed]
- Bundy, A.L.; Saltzman, D.H.; Pober, B.; Fine, C.; Emerson, D.; Doubilet, P.M. Antenatal sonographic findings in trisomy 18. J. Ultrasound Med. 1986, 5, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Cook, J.P.; Mehta, L. Changing demography of trisomy 18. Arch. Dis. Child. 1986, 61, 1035–1036. [Google Scholar] [CrossRef] [PubMed]
- Root, S.; Carey, J.C. Survival in trisomy 18. Am. J. Med. Genet. 1994, 49, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Naguib, K.K.; Al-Awadi, S.A.; Bastaki, L.; Moussa, M.A.; Abulhassan, S.A.; Tayel, S.; Murthy, K. Clustering of trisomy 18 in Kuwait: Genetic predisposition or environmental? Ann. Saudi Med. 1999, 19, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, G.E.; Douglass, E.C.; Pollock, B.H.; Finegold, M.J.; Schneider, N.R. Cytogenetic evaluation of a large series of hepatoblastomas: Numerical abnormalities with recurring aberrations involving 1q12-q21. Genes Chromosomes Cancer 2005, 44, 177–184. [Google Scholar] [CrossRef]
- Farmakis, S.G.; Barnes, A.M.; Carey, J.C.; Braddock, S.R. Solid tumor screening recommendations in trisomy 18. Am. J. Med. Genet. A 2019, 179, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Mamlok, V.; Nichols, M.; Lockhart, L.; Mamlok, R. Trisomy 18 and hepatoblastoma. Am. J. Med. Genet. 1989, 33, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Teraguchi, M.; Nogi, S.; Ikemoto, Y.; Ogino, H.; Kohdera, U.; Sakaida, N.; Okamura, A.; Hamada, Y.; Kobayashi, Y. Multiple hepatoblastomas associated with trisomy 18 in a 3-year-old girl. Pediatr. Hematol. Oncol. 1997, 14, 463–467. [Google Scholar] [CrossRef]
- Dasouki, M.; Barr, M., Jr. Trisomy 18 and hepatic neoplasia. Am. J. Med. Genet. 1987, 27, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Uemoto, S.; Asonuma, K.; Katayama, T.; Utsunomiya, H.; Akiyama, Y.; Sasaki, M.S.; Ozawa, K. Hepatoblastoma in a 2-year-old girl with trisomy 18. Eur. J. Pediatr. Surg. 1992, 2, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Bove, K.E.; Soukup, S.; Ballard, E.T.; Ryckman, F. Hepatoblastoma in a child with trisomy 18: Cytogenetics, liver anomalies, and literature review. Pediatr. Pathol. Lab. Med. 1996, 16, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Ikeda, H.; Koizumi, T. Hepatoblastoma associated with trisomy 18 syndrome: A case report and a review of the literature. Pediatr. Int. 2001, 43, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, K.S.; Baum, R.; Fung, B.; Yeager, N.; Leonis, M.A.; Wagner, L.M.; Tiao, G.; Ross, M.E. Chemoresistant hepatoblastoma in a patient with mosaic trisomy 18 treated with orthotopic liver transplantation. Pediatr. Blood Cancer 2011, 56, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.H.; Lai, A.; Chen, C.K.; Chang, K.T.; Tan, A.M. Association of trisomy 18 with hepatoblastoma and its implications. Eur. J. Pediatr. 2014, 173, 1595–1598. [Google Scholar] [CrossRef] [PubMed]
- Uekusa, S.; Sugito, K.; Kawashima, H.; Yoshizawa, S.; Furuya, T.; Ohashi, K.; Ikeda, T.; Koshinaga, T.; Mugishima, H. Successful treatment for hepatoblastoma in a 1-year-old boy with trisomy 18. Pediatr. Int. 2012, 54, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Suzuki, R.; Urabe, K.; Kawamura, Y.; Masuda, M.; Kishi, K.; Takitani, K.; Katayama, H.; Tomiyama, H.; Hayashi, M.; et al. Therapeutic experience with hepatoblastoma associated with trisomy 18. Pediatr. Blood Cancer 2018, 65, e27093. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.J.; Rubinstein, J.; Gosain, A.; Tiao, G.; Head, T.; Pratap, J.N.; Williams, R.; Helmig, S.; Geller, J.; Langham, M.; et al. Surgical and anesthetic management for hepatectomy in two pediatric patients with trisomy 18, pulmonary hypertension, and hepatoblastoma. Pediatr. Blood Cancer 2019, 66, e27678. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Wu, T.C. A Long-Term Survivor of Trisomy 18. Cureus 2024, 16, e51491. [Google Scholar] [CrossRef] [PubMed]
- Shirane, K.; Yoshimi, A.; Masuko, T.; Kajikawa, D.; Toma, M.; Idesawa, H.; Tsukada, Y.; Yano, Y.; Kato, K.; Motoyama, K.; et al. Successful Treatment for Hepatoblastoma in Trisomy 18: A Case Report. J. Pediatr. Hematol. Oncol. 2024, 46, e83–e86. [Google Scholar] [CrossRef] [PubMed]
- Valentin, L.I.; Perez, L.; Masand, P. Hepatoblastoma Associated with Trisomy 18. J. Pediatr. Genet. 2015, 4, 204–206. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Trisomy 18 | |
|---|---|
| Total | n = 18 |
| Mosaic | n = 4 |
| Gender | |
| Female | n = 17 |
| Male | n = 5 |
| Age at the moment of hepatoblastoma diagnosis | |
| 0–6 months | n = 5 |
| 7–12 months | n = 9 |
| 1–2 years | n = 5 |
| 2–4 years | n = 3 |
| above 4 years | n = 0 |
| Treatment | |
| Surgery | n = 5 |
| Chemotherapy | n = 0 |
| Surgery + Chemotherapy | n =12 |
| Surgery + Chem + Liver transplant | n = 1 |
| No treatment | n = 4 |
| Disease status | |
| remission | n = 16 |
| progression | n = 6 |
| Death | |
| due to cancer | n = 2 |
| due to organ failure | n = 3 |
| related to 18 trisomy | n = 1 |
| no reported death | n = 16 |
| Age of delivery | |
| preterm | n = 9 |
| at term | n = 11 |
| n/a | n = 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sosnowska-Sienkiewicz, P.; Kamińska, A.; Anderko, I.; Telman-Kołodziejczyk, G.; Mańkowski, P.; Januszkiewicz-Lewandowska, D. Therapeutic Management and Outcomes of Hepatoblastoma in a Pediatric Patient with Mosaic Edwards Syndrome. Genes 2024, 15, 463. https://doi.org/10.3390/genes15040463
Sosnowska-Sienkiewicz P, Kamińska A, Anderko I, Telman-Kołodziejczyk G, Mańkowski P, Januszkiewicz-Lewandowska D. Therapeutic Management and Outcomes of Hepatoblastoma in a Pediatric Patient with Mosaic Edwards Syndrome. Genes. 2024; 15(4):463. https://doi.org/10.3390/genes15040463
Chicago/Turabian StyleSosnowska-Sienkiewicz, Patrycja, Alicja Kamińska, Iwona Anderko, Gabriela Telman-Kołodziejczyk, Przemysław Mańkowski, and Danuta Januszkiewicz-Lewandowska. 2024. "Therapeutic Management and Outcomes of Hepatoblastoma in a Pediatric Patient with Mosaic Edwards Syndrome" Genes 15, no. 4: 463. https://doi.org/10.3390/genes15040463
APA StyleSosnowska-Sienkiewicz, P., Kamińska, A., Anderko, I., Telman-Kołodziejczyk, G., Mańkowski, P., & Januszkiewicz-Lewandowska, D. (2024). Therapeutic Management and Outcomes of Hepatoblastoma in a Pediatric Patient with Mosaic Edwards Syndrome. Genes, 15(4), 463. https://doi.org/10.3390/genes15040463