
Heritability of Gene Expression Measured from Peripheral Blood in Older Adults


 , , , and
, , , and 
Abstract
:1. Introduction
2. Methods
2.1. Participants
2.2. Gene Expression Analysis
2.3. Statistical Analysis and Bioinformatics
2.3.1. Heritability
2.3.2. Gene Set Analysis
2.3.3. Correlations between Gene Expression Heritability with Gene Length and Percentage GC Content
2.3.4. Ageing and Longevity Enrichment Analysis
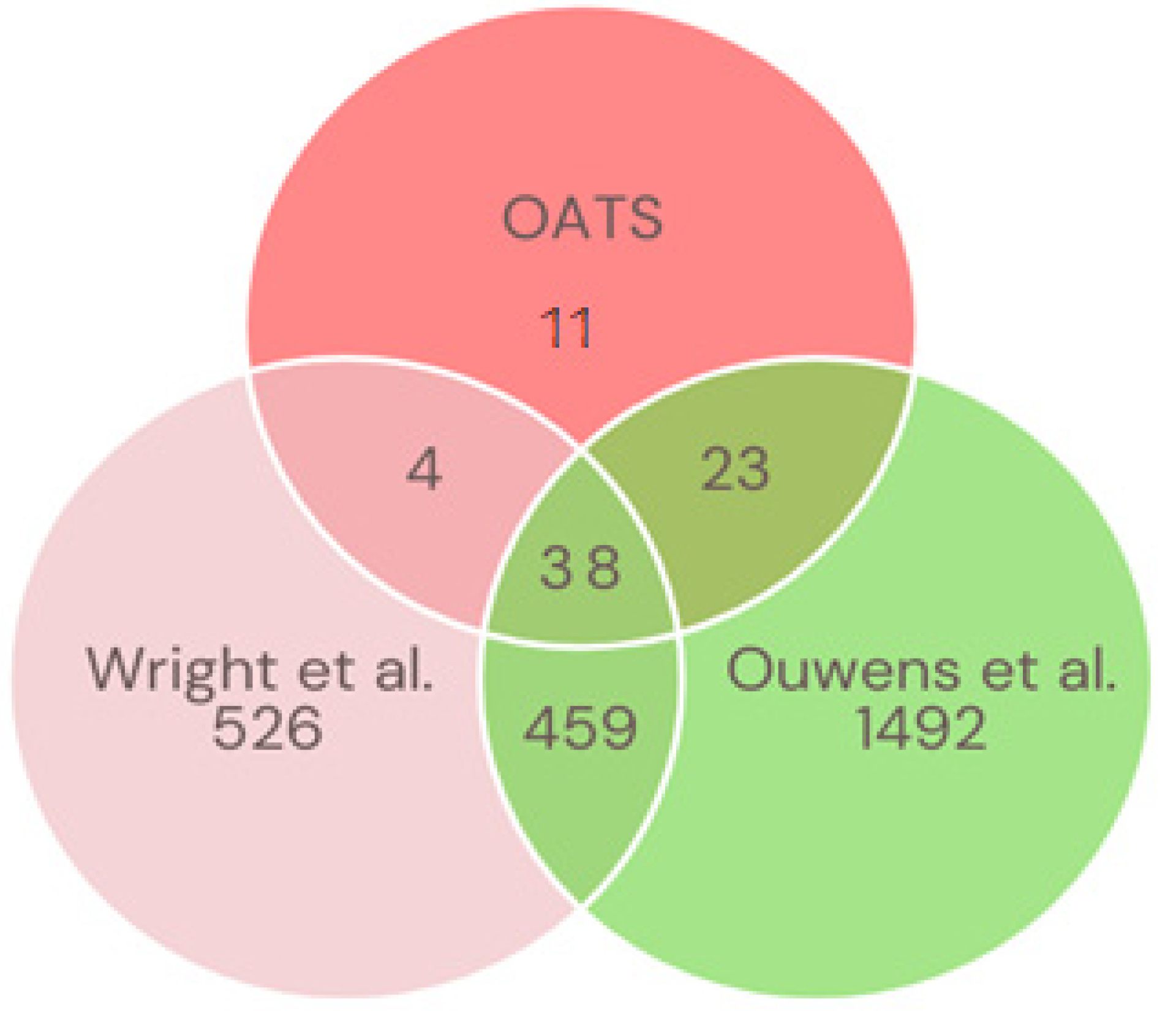
2.3.5. Overlap Analysis for the Heritability of Gene Expression across Three Studies
3. Results
3.1. Demographics
3.2. Heritability Analysis
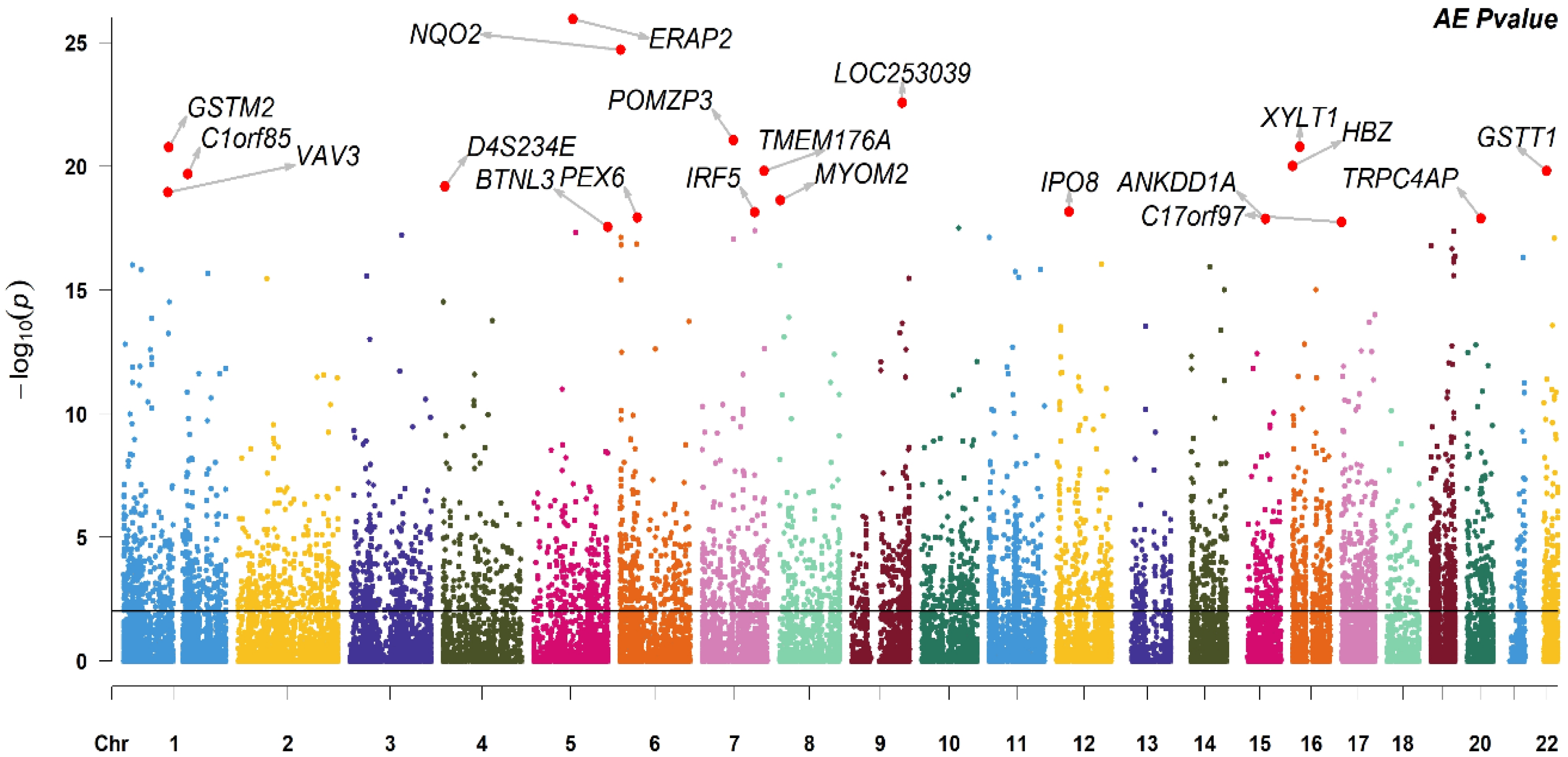
3.2.1. Probe Heritability
3.2.2. Gene Heritability
3.3. Correlations between Gene Expression Heritability with Gene Length and Percentage GC Content
3.4. Gene Set Analysis
3.5. Ageing and Longevity Enrichment Analysis
3.6. Candidate Non-Heritable Genes
3.7. Cross-Study Comparison of Gene Expression Heritability across Three Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grundberg, E.; Small, K.S.; Hedman, Å.K.; Nica, A.C.; Buil, A.; Keildson, S.; Bell, J.T.; Yang, T.P.; Meduri, E.; Barrett, A.; et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat. Genet. 2012, 44, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Morley, M.; Molony, C.M.; Weber, T.M.; Devlin, J.L.; Ewens, K.G.; Spielman, R.S.; Cheung, V.G. Genetic analysis of genome-wide variation in human gene expression. Nature 2004, 430, 743–747. [Google Scholar] [CrossRef]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Ouwens, K.G.; Jansen, R.; Nivard, M.G.; van Dongen, J.; Frieser, M.J.; Hottenga, J.J.; Arindrarto, W.; Claringbould, A.; van Iterson, M.; Mei, H.; et al. A characterization of cis- and trans-heritability of RNA-Seq-based gene expression. Eur. J. Hum. Genet. EJHG 2020, 28, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.E.; Henders, A.K.; McRae, A.F.; Wright, M.J.; Martin, N.G.; Dermitzakis, E.T.; Montgomery, G.W.; Visscher, P.M. Genetic control of gene expression in whole blood and lymphoblastoid cell lines is largely independent. Genome Res. 2012, 22, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Helgason, A.; Thorleifsson, G.; McCarroll, S.A.; Kong, A.; Stefansson, K. Single-tissue and cross-tissue heritability of gene expression via identity-by-descent in related or unrelated individuals. PLoS Genet. 2011, 7, e1001317. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.A.; Sullivan, P.F.; Brooks, A.I.; Zou, F.; Sun, W.; Xia, K.; Madar, V.; Jansen, R.; Chung, W.; Zhou, Y.H.; et al. Heritability and genomics of gene expression in peripheral blood. Nat. Genet. 2014, 46, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, L.R.; Holloway, A.; McRae, A.; Yang, J.; Small, K.; Zhao, J.; Zeng, B.; Bakshi, A.; Metspalu, A.; Dermitzakis, M.; et al. The Genetic Architecture of Gene Expression in Peripheral Blood. Am. J. Hum. Genet. 2017, 100, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Christensen, K.; Christiansen, L.; Frederiksen, H.; Bathum, L.; Dahlgaard, J.; Kruse, T.A. Genetic dissection of gene expression observed in whole blood samples of elderly Danish twins. Hum. Genet. 2005, 117, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, P.S.; Lammel, A.; Trollor, J.N.; Lee, T.; Wright, M.J.; Ames, D.; Wen, W.; Martin, N.G.; Brodaty, H.; Schofield, P.R. A comprehensive neuropsychiatric study of elderly twins: The Older Australian Twins Study. Twin Res. Hum. Genet. Off. J. Int. Soc. Twin Stud. 2009, 12, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Batouli, S.A.; Sachdev, P.S.; Wen, W.; Wright, M.J.; Suo, C.; Ames, D.; Trollor, J.N. The heritability of brain metabolites on proton magnetic resonance spectroscopy in older individuals. NeuroImage 2012, 62, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Gallego Romero, I.; Pai, A.A.; Tung, J.; Gilad, Y. RNA-seq: Impact of RNA degradation on transcript quantification. BMC Biol. 2014, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Ciobanu, L.G.; Sachdev, P.S.; Trollor, J.N.; Reppermund, S.; Thalamuthu, A.; Mather, K.A.; Cohen-Woods, S.; Stacey, D.; Toben, C.; Schubert, K.O.; et al. Co-expression network analysis of peripheral blood transcriptome identifies dysregulated protein processing in endoplasmic reticulum and immune response in recurrent MDD in older adults. J. Psychiatr. Res. 2018, 107, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.; Slagboom, P.E.; Draisma, H.H.; Martin, N.G.; Boomsma, D.I. The continuing value of twin studies in the omics era. Nat. Rev. Genet. 2012, 13, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Neale, M.C.; Hunter, M.D.; Pritikin, J.N.; Zahery, M.; Brick, T.R.; Kirkpatrick, R.M.; Estabrook, R.; Bates, T.C.; Maes, H.H.; Boker, S.M. OpenMx 2.0: Extended Structural Equation and Statistical Modeling. Psychometrika 2016, 81, 535–549. [Google Scholar] [CrossRef]
- Pritikin, J.N.; Hunter, M.D.; Boker, S. Modular Open-Source Software for Item Factor Analysis. Educ. Psychol. Meas. 2015, 75, 458–474. [Google Scholar] [CrossRef] [PubMed]
- McCaw, Z.R.; Lane, J.M.; Saxena, R.; Redline, S.; Lin, X. Operating characteristics of the rank-based inverse normal transformation for quantitative trait analysis in genome-wide association studies. Biometrics 2020, 76, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef] [PubMed]
- De Magalhães, J.P.; Curado, J.; Church, G.M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 2009, 25, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.W.; Thalamuthu, A.; Braidy, N.; Mather, K.A.; Liu, Y.; Ciobanu, L.; Baune, B.T.; Armstrong, N.J.; Kwok, J.; Schofield, P.; et al. Genetic and environmental determinants of variation in the plasma lipidome of older Australian twins. eLife 2020, 9, e58954. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Lipnicki, D.M.; Crawford, J.D.; Henry, J.D.; Trollor, J.N.; Ames, D.; Wright, M.J.; Sachdev, P.S. Leisure activity, health, and medical correlates of neurocognitive performance among monozygotic twins: The Older Australian Twins Study. J. Gerontol. Ser. B Psychol. Sci. Soc. Sci. 2014, 69, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Hearn, A.; York, I.A.; Rock, K.L. The specificity of trimming of MHC class I-presented peptides in the endoplasmic reticulum. J. Immunol. 2009, 183, 5526–5536. [Google Scholar] [CrossRef] [PubMed]
- Lorente, E.; Redondo-Antón, J.; Martín-Esteban, A.; Guasp, P.; Barnea, E.; Lauzurica, P.; Admon, A.; López de Castro, J.A. Substantial Influence of ERAP2 on the HLA-B*40:02 Peptidome: Implications for HLA-B*27-Negative Ankylosing Spondylitis. Mol. Cell Proteom. 2019, 18, 2298–2309. [Google Scholar] [CrossRef] [PubMed]
- Voronin, M.V.; Kadnikov, I.A.; Zainullina, L.F.; Logvinov, I.O.; Verbovaya, E.R.; Antipova, T.A.; Vakhitova, Y.V.; Seredenin, S.B. Neuroprotective Properties of Quinone Reductase 2 Inhibitor M-11, a 2-Mercaptobenzimidazole Derivative. Int. J. Mol. Sci. 2021, 22, 13061. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Q.; Zhu, S.Y.; He, Y.; Yu, K.D. Association Between a Tri-allelic Polymorphism in the Estrogen Metabolism Oxidoreductase NRH:Quinone Oxidoreductase 2 Gene and Risk of Breast Cancer by Molecular Subtype. Front. Genet. 2021, 12, 658285. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.R.; Bhatia, G.; Gusev, A.; Finucane, H.K.; Bulik-Sullivan, B.K.; Pollack, S.J.; de Candia, T.R.; Lee, S.H.; Wray, N.R.; Kendler, K.S.; et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 2015, 47, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Gelfman, S.; Cohen, N.; Yearim, A.; Ast, G. DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res. 2013, 23, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Polak, P.; Nemesh, J.; Michaelson, J.J.; Sebat, J.; Sunyaev, S.R.; McCarroll, S.A. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet. 2012, 91, 1033–1040. [Google Scholar] [CrossRef]
- Daunay, A.; Hardy, L.M.; Bouyacoub, Y.; Sahbatou, M.; Touvier, M.; Blanché, H.; Deleuze, J.F.; How-Kit, A. Centenarians consistently present a younger epigenetic age than their chronological age with four epigenetic clocks based on a small number of CpG sites. Aging 2022, 14, 7718–7733. [Google Scholar] [CrossRef] [PubMed]
- Dec, E.; Clement, J.; Cheng, K.; Church, G.M.; Fossel, M.B.; Rehkopf, D.H.; Rosero-Bixby, L.; Kobor, M.S.; Lin, D.T.; Lu, A.T.; et al. Centenarian clocks: Epigenetic clocks for validating claims of exceptional longevity. Geroscience 2023, 45, 1817–1835. [Google Scholar] [CrossRef] [PubMed]
- Napolioni, V.; Giannì, P.; Carpi, F.M.; Concetti, F.; Lucarini, N. Haptoglobin (HP) polymorphisms and human longevity: A cross-sectional association study in a Central Italy population. Clin. Chim. Acta 2011, 412, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Tindale, L.C.; Leach, S.; Spinelli, J.J.; Brooks-Wilson, A.R. Lipid and Alzheimer’s disease genes associated with healthy aging and longevity in healthy oldest-old. Oncotarget 2017, 8, 20612–20621. [Google Scholar] [CrossRef] [PubMed]
- Mirauta, B.A.; Seaton, D.D.; Bensaddek, D.; Brenes, A.; Bonder, M.J.; Kilpinen, H.; Stegle, O.; Lamond, A.I. Population-scale proteome variation in human induced pluripotent stem cells. eLife 2020, 9, e57390. [Google Scholar] [CrossRef] [PubMed]
- García-García, G.; Sanchez-Navarro, I.; Aller, E.; Jaijo, T.; Fuster-Garcia, C.; Rodríguez-Munoz, A.; Vallejo, E.; Tellería, J.J.; Vázquez, S.; Beltrán, S.; et al. Exome sequencing identifies PEX6 mutations in three cases diagnosed with Retinitis Pigmentosa and hearing impairment. Mol. Vis. 2020, 26, 216–225. [Google Scholar] [PubMed]
- Schieferdecker, A.; Wendler, P. Structural Mapping of Missense Mutations in the Pex1/Pex6 Complex. Int. J. Mol. Sci. 2019, 20, 3756. [Google Scholar] [CrossRef] [PubMed]
- Viñuela, A.; Brown, A.A.; Buil, A.; Tsai, P.C.; Davies, M.N.; Bell, J.T.; Dermitzakis, E.T.; Spector, T.D.; Small, K.S. Age-dependent changes in mean and variance of gene expression across tissues in a twin cohort. Hum. Mol. Genet. 2018, 27, 732–741. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variable | Total Sample | MZ | DZ | p-Value |
|---|---|---|---|---|
| N | 246 | 142 (71 pairs) | 104 (52 pairs) | N/A |
| Sex—Female, N (%) | 172 (69.91) | 102 (71.83) | 70 (67.31) | 0.402 |
| Age (yrs) Mean (SD) | 75.79 (5.44) | 75.60 (5.49) | 76.04 (5.39) | 0.537 |
| Age Range (yrs) | (69.4–93.5) | (69.7–93.5) | (69.4–93.1) | N/A |
| BMI (Kg/m2) | 27.63 (4.74) | 27.92 (4.89) | 27.22 (4.52) | 0.764 |
| WBC count (K/μL) | 6.41 (1.66) | 6.33 (1.59) | 6.51 (1.73) | 0.392 |
| Current smoker | 8 (3.25%) | 5 | 3 | NA |
| Alcohol user (>30 drinks per month) | 77 (31.30%) | 46 | 31 | NA |
| Probe ID | Gene Symbol | Chr | h2 | CI | Bootstrap CI | FDR |
|---|---|---|---|---|---|---|
| ILMN_1743145 | ERAP2 | 5 | 0.87 | 0.81–0.91 | 0.84–0.90 | 2.91 × 10−22 |
| ILMN_1712918 | NQO2 | 6 | 0.86 | 0.80–0.91 | 0.82–0.91 | 2.54 × 10−21 |
| ILMN_3236498 | LOC253039 | 9 | 0.85 | 0.78–0.90 | 0.81–0.89 | 2.35 × 10−19 |
| ILMN_1805377 | POMZP3 | 7 | 0.81 | 0.73–0.87 | 0.76–0.87 | 5.81 × 10−18 |
| ILMN_1830462 | XYLT1 | 16 | 0.85 | 0.77–0.90 | 0.80–0.89 | 7.66 × 10−18 |
| ILMN_2201580 | GSTM2 | 1 | 0.8 | 0.71–0.86 | 0.74–0.87 | 7.66 × 10−18 |
| ILMN_1713458 | HBZ | 16 | 0.78 | 0.69–0.85 | 0.73–0.84 | 3.74 × 10−17 |
| ILMN_1791511 | TMEM176A | 7 | 0.81 | 0.72–0.87 | 0.76–0.86 | 4.58 × 10−17 |
| ILMN_1730054 | GSTT1 | 22 | 0.8 | 0.71–0.86 | 0.75–0.86 | 4.58 × 10−17 |
| ILMN_1698243 | C1orf85 | 1 | 0.8 | 0.71–0.86 | 0.75–0.84 | 5.45 × 10−17 |
| GO Biological Processes/Canonical Pathways | N Genes in Pathway | N Genes Overlap | FDR |
|---|---|---|---|
| REACTOME_NEUTROPHIL_DEGRANULATION | 389 | 51 | 7.82 × 10−16 |
| REACTOME_INNATE_IMMUNE_SYSTEM | 826 | 66 | 2.17 × 10−10 |
| REACTOME_ANTIMICROBIAL_PEPTIDES | 56 | 13 | 7.59 × 10−6 |
| GOBP_ANTIMICROBIAL_HUMORAL_RESPONSE | 80 | 16 | 1.10 × 10−5 |
| GOBP_IMMUNE_RESPONSE | 1237 | 73 | 3.81 × 10−5 |
| GOBP_ANTIMICROBIAL_HUMORAL_IMMUNE_RESPONSE_MEDIATED_BY_ANTIMICROBIAL_PEPTIDE | 53 | 12 | 9.82 × 10−5 |
| GOBP_DEFENSE_RESPONSE | 1297 | 73 | 1.27 × 10−4 |
| GOBP_DEFENSE_RESPONSE_TO_BACTERIUM | 212 | 23 | 1.27 × 10−4 |
| GOBP_INFLAMMATORY_RESPONSE | 645 | 45 | 1.27 × 10−4 |
| GOBP_HUMORAL_IMMUNE_RESPONSE | 168 | 19 | 7.2 × 10−4 |
| Gene Symbol | Probe ID | Chr | OATS | Wright | Ouwens | |||
|---|---|---|---|---|---|---|---|---|
| h2 | FDR | h2 | FDR | h2 | FDR | |||
| ERAP2 | ENSG00000164308 | 5 | 0.87 | 8.94 × 10−4 | 0.88 | 3.53 × 10−25 | 0.85 | 1.95 × 10−28 |
| NQO2 | ENSG00000124588 | 6 | 0.86 | 4.64 × 10−5 | 0.59 | 3.21 × 10−8 | 0.87 | 2.25 × 10−37 |
| XYLT1 | ENSG00000103489 | 16 | 0.85 | 3.14 × 10−5 | 0.41 | 6.56 × 10−3 | 0.43 | 2.58 × 10−3 |
| PEX6 | ENSG00000124587 | 6 | 0.83 | 1.32 × 10−5 | 0.32 | 3.21 × 10−2 | 0.88 | 1.19 × 10−40 |
| TMEM176A | ENSG00000002933 | 7 | 0.81 | 7.35 × 10−3 | 0.61 | 2.50 × 10−13 | 0.91 | 3.76 × 10−45 |
| LILRA3 | ENSG00000170866 | 19 | 0.81 | 4.64 × 10−5 | 0.75 | 8.76 × 10−15 | 0.85 | 1.08 × 10−25 |
| NSG1 (D4S234E) | ENSG00000168824 | 4 | 0.81 | 1.50 × 10−4 | 0.35 | 1.33 × 10−2 | 0.82 | 5.02 × 10−24 |
| ANKDD1A | ENSG00000166839 | 15 | 0.80 | 8.94 × 10−4 | 0.34 | 2.59 × 10−2 | 0.43 | 7.75 × 10−4 |
| CFD | ENSG00000197766 | 19 | 0.80 | 1.85 × 10−4 | 0.57 | 1.23 × 10−9 | 0.74 | 6.82 × 10−11 |
| MYOM2 | ENSG00000036448 | 8 | 0.79 | 4.80 × 10−3 | 0.72 | 3.01 × 10−20 | 0.87 | 1.20 × 10−32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanchibhotla, S.C.; Mather, K.A.; Armstrong, N.J.; Ciobanu, L.G.; Baune, B.T.; Catts, V.S.; Schofield, P.R.; Trollor, J.N.; Ames, D.; Sachdev, P.S.; et al. Heritability of Gene Expression Measured from Peripheral Blood in Older Adults. Genes 2024, 15, 495. https://doi.org/10.3390/genes15040495
Kanchibhotla SC, Mather KA, Armstrong NJ, Ciobanu LG, Baune BT, Catts VS, Schofield PR, Trollor JN, Ames D, Sachdev PS, et al. Heritability of Gene Expression Measured from Peripheral Blood in Older Adults. Genes. 2024; 15(4):495. https://doi.org/10.3390/genes15040495
Chicago/Turabian StyleKanchibhotla, Sri C., Karen A. Mather, Nicola J. Armstrong, Liliana G. Ciobanu, Bernhard T. Baune, Vibeke S. Catts, Peter R. Schofield, Julian N. Trollor, David Ames, Perminder S. Sachdev, and et al. 2024. "Heritability of Gene Expression Measured from Peripheral Blood in Older Adults" Genes 15, no. 4: 495. https://doi.org/10.3390/genes15040495
APA StyleKanchibhotla, S. C., Mather, K. A., Armstrong, N. J., Ciobanu, L. G., Baune, B. T., Catts, V. S., Schofield, P. R., Trollor, J. N., Ames, D., Sachdev, P. S., & Thalamuthu, A. (2024). Heritability of Gene Expression Measured from Peripheral Blood in Older Adults. Genes, 15(4), 495. https://doi.org/10.3390/genes15040495