
A TMEM63A Nonsense Heterozygous Variant Linked to Infantile Transient Hypomyelinating Leukodystrophy Type 19?


 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Data Collection
2.2. DNA Sequence Analysis
3. Results
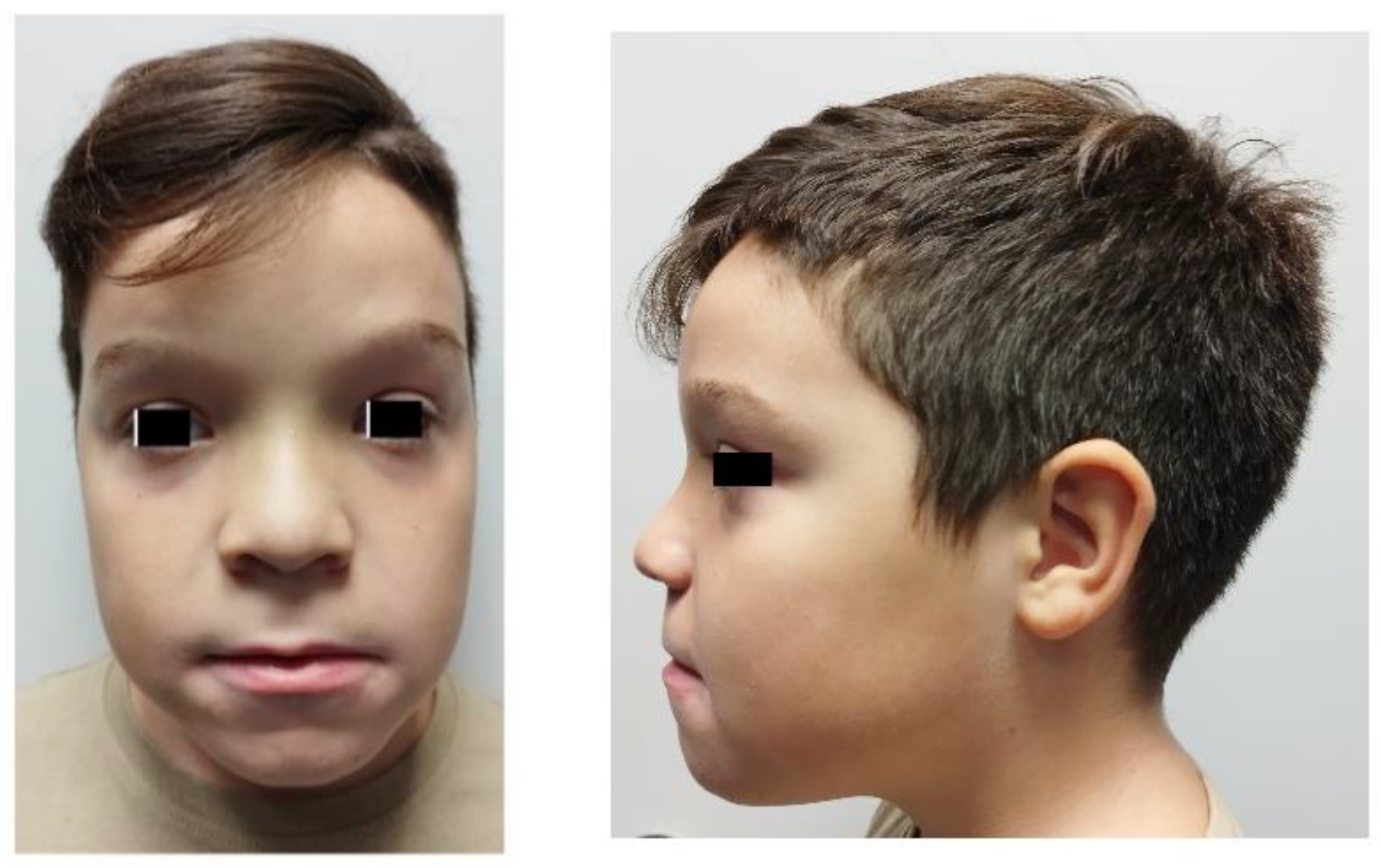
3.1. Case Report
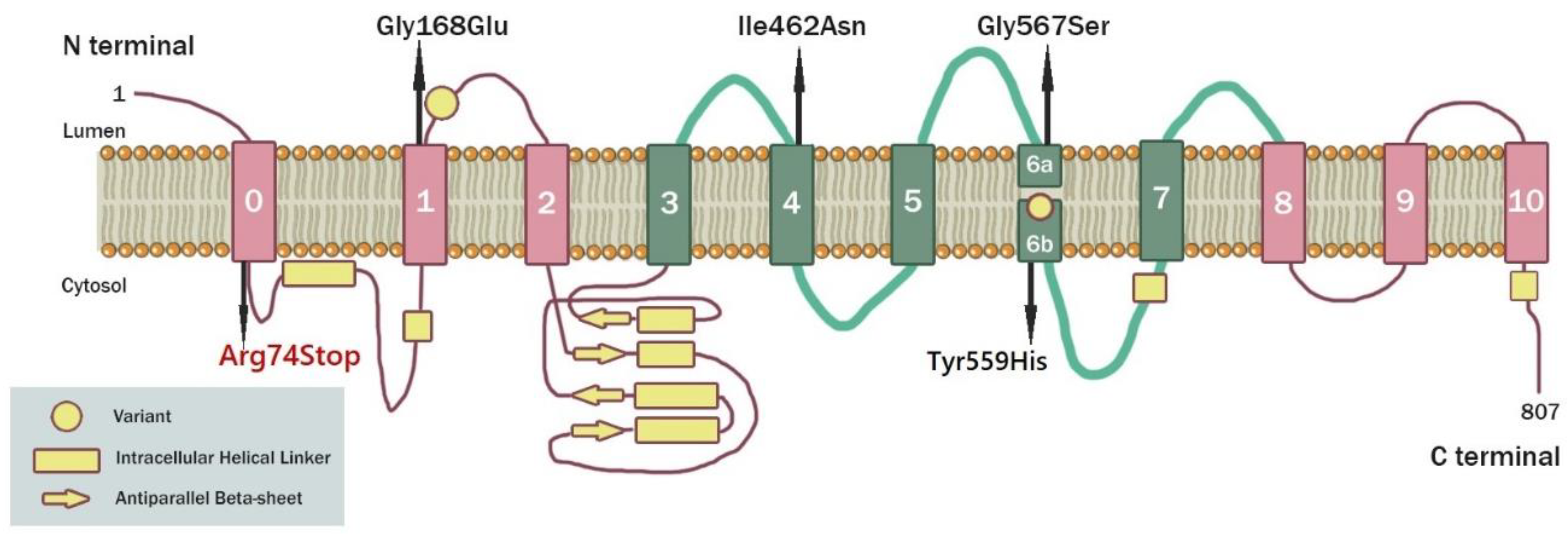
3.2. Molecular Genetic Analysis
3.3. Follow-Up
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van der Knaap, M.S.; Schiffmann, R.; Mochel, F.; Wolf, N.I. Diagnosis, prognosis, and treatment of leukodystrophies. Lancet Neurol. 2019, 18, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Ffrench-Constant, C.; van der Knaap, M.S. Hypomyelinating leukodystrophies-unravelling myelin biology. Nat. Rev. Neurol. 2021, 17, 88–103. [Google Scholar] [CrossRef]
- Tomassy, G.S.; Dershowitz, L.B.; Arlotta, P. Diversity Matters: A Revised Guide to Myelination. Trends Cell Biol. 2016, 26, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Vanderver, A.; Hussey, H.; Schmidt, J.L.; Pastor, W.; Hoffman, H.J. Relative incidence of inherited white matter disorders in childhood to acquired pediatric demyelinating disorders. Semin. Pediatr. Neurol. 2012, 19, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Chanoumidou, K.; Mozafari, S.; Baron-Van Evercooren, A.; Kuhlmann, T. Stem cell derived oligodendrocytes to study myelin diseases. Glia 2020, 68, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Amado, D.A.; Davidson, B.L. Gene therapy for ALS: A review. Mol. Ther. 2021, 29, 3345–3358. [Google Scholar] [CrossRef]
- Meschkat, M.; Steyer, A.M.; Weil, M.T.; Kusch, K.; Jahn, O.; Piepkorn, L.; Agüi-Gonzalez, P.; Phan, N.T.N.; Ruhwedel, T.; Sadowski, B.; et al. White matter integrity in mice requires continuous myelin synthesis at the inner tongue. Nat. Commun. 2022, 13, 1163. [Google Scholar] [CrossRef]
- Kassmann, C.M. Myelin peroxisomes-essential organelles for the maintenance of white matter in the nervous system. Biochimie 2014, 98, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Cali, J.J.; Hsieh, C.L.; Francke, U.; Russell, D.W. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J. Biol. Chem. 1991, 266, 7779–7783. [Google Scholar] [CrossRef]
- Weigel, M.; Wang, L.; Fu, M.M. Microtubule organization and dynamics in oligodendrocytes, astrocytes, and microglia. Dev. Neurobiol. 2021, 81, 310–320. [Google Scholar] [CrossRef]
- Merheb, E.; Cui, M.H.; DuBois, J.C.; Branch, C.A.; Gulinello, M.; Shafit-Zagardo, B.; Moir, R.D.; Willis, I.M. Defective myelination in an RNA polymerase III mutant leukodystrophic mouse. Proc. Natl. Acad. Sci. USA 2021, 118, e2024378118. [Google Scholar] [CrossRef] [PubMed]
- Meservey, L.M.; Topkar, V.V.; Fu, M.M. mRNA transport and local translation in glia. Trends Cell Biol. 2021, 31, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.I.; Gutierrez Salazar, M.; Guerrero, K.; Thiffault, I.; Salomons, G.S.; Gauquelin, L.; Tran, L.T.; Forget, D.; Gauthier, M.-S.; Waisfisz, Q.; et al. Bi-allelic mutations in EPRS, encoding the glutamyl-prolyl-aminoacyl-tRNA synthetase, cause a hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2018, 102, 676–684. [Google Scholar] [CrossRef]
- Van der Knaap, M.S.; Boor, I.; Estévez, R. Megalencephalic leukoencephalopathy with subcortical cysts: Chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet. Neurol. 2012, 11, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Bourre, J.M.; Jacque, C.; Nguyen-Legros, J.; Bornhofen, J.H.; Araoz, C.A.; Daudu, O.; Baumann, N. Pelizaeus-merzbacher disease: Biochemical analysis of isolated myelin (electron-microscopy; protein, lipid and unsubstituted fatty acids analysis). Eur. Neurol. 1978, 17, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Yapijakis, C.; Kalogera, S.; Angelopoulou, A.; Paraskevas, G.; Kapaki, E. Craniofacial and neurological phenotype in a case of oculodentodigital syndrome. Adv. Exp. Med. Biol. 2021, 1339, 325–329. [Google Scholar] [PubMed]
- Elpidorou, M.; Poulter, J.A.; Szymanska, K.; Baron, W.; Junger, K.; Boldt, K.; Ueffing, M.; Green, L.; Livingston, J.H.; Sheridan, E.G.; et al. Missense mutation of MAL causes a rare leukodystrophy similar to Pelizaeus-Merzbacher disease. Eur. J. Hum. Genet. 2022, 30, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Woodward, L.J.; Anderson, P.J.; Austin, N.C.; Howard, K.; Inder, T.E. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N. Engl. J. Med. 2006, 355, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Marelli, A.J.; Mackie, A.S.; Ionescu-Ittu, R.; Rahme, E.; Pilote, L. Congenital heart disease in the general population: Changing prevalence and age distribution. Circulation 2007, 115, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar] [CrossRef]
- Stoll, B.J.; Hansen, N.I.; Adams-Chapman, I.; Fanaroff, A.A.; Hintz, S.R.; Vohr, B.; Higgins, R.D.; National Institute of Child Health and Human Development Neonatal Research Network. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. JAMA 2004, 292, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Helman, G.; Murthy, S.E.; Ji, H.; Crawford, J.; Kubisiak, T.; Bent, S.J.; Xiao, J.; Taft, R.J.; Coombs, A.; et al. Heterozygous variants in the mechanosensitive ion channel TMEM63A result in transient hypomyelination of infancy. Am. J. Hum. Genet. 2019, 105, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Tonduti, D.; Mura, E.; Masnada, S.; Bertini, E.; Aiello, C.; Zini, D.; Parmeggiani, L.; Cantalupo, G.; Talenti, G.; Veggiotti, P.; et al. Spinal cord involvement and paroxysmal events in ‘infantile onset transient hypomyelination’ due to TMEM63A mutation. J. Hum. Genet. 2021, 66, 1035–1037. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Ji, H.; Kubisiak, T.; Wu, Y.; Xiao, J.; Gu, Q.; Yang, Y.; Xie, H.; Ji, T.; Gao, K.; et al. Genetic analysis of 20 patients with hypomyelinating leukodystrophy by trio-based whole-exome sequencing. J. Hum. Genet. 2021, 66, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Vanderver, A.; van Spaendonk, R.M.; Schiffmann, R.; Brais, B.; Bugiani, M.; Sistermans, E.; Catsman-Berrevoets, C.; Kros, J.M.; Pinto, P.S.; et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014, 83, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Hobson, G.M.; Garbern, J.Y. Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders. Semin. Neurol. 2012, 32, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.E.; Dubin, A.E.; Whitwam, T.; Jojoa-Cruz, S.; Caha-lan, S.M.; Mousavi SA, R.; Ward, A.B.; Patapoutian, A. OSCA/TMEM63 are an Evolutionarily Conserved Family of Mechanically Activated Ion Channels. eLife 2018, 7, e41844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yan, X.; Liu, Y.; Zhang, P.; Ni, X. Co-expression of mouse TMEM63A, TMEM63B and TMEM63C confers hyperosmolarity activated ion currents in HEK293 cells. Cell Biochem. Funct. 2016, 34, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Rawson, S.; Shen, Z.; Tamilselvan, E.; Smith, H.E.; Halford, J.; Shen, C.; Murthy, S.E.; Ulbrich, M.H.; Sotomayor, M.; et al. TMEM63 proteins function as monomeric high-threshold mechanosensitive ion channels. Neuron 2023, 111, 3195–3210.e7. [Google Scholar] [CrossRef]
- Chen, X.; Wang, N.; Liu, J.-W.; Zeng, B.; Chen, G.-L. TMEM63 mechanosensitive ion channels: Activation mechanisms, biological functions and human genetic disorders. Biochem. Biophys. Res. Commun. 2023, 683, 149111. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siori, D.; Vlachakis, D.; Makrythanasis, P.; Traeger-Synodinos, J.; Veltra, D.; Kampouraki, A.; Chrousos, G.P. A TMEM63A Nonsense Heterozygous Variant Linked to Infantile Transient Hypomyelinating Leukodystrophy Type 19? Genes 2024, 15, 525. https://doi.org/10.3390/genes15050525
Siori D, Vlachakis D, Makrythanasis P, Traeger-Synodinos J, Veltra D, Kampouraki A, Chrousos GP. A TMEM63A Nonsense Heterozygous Variant Linked to Infantile Transient Hypomyelinating Leukodystrophy Type 19? Genes. 2024; 15(5):525. https://doi.org/10.3390/genes15050525
Chicago/Turabian StyleSiori, Dimitra, Dimitrios Vlachakis, Periklis Makrythanasis, Joanne Traeger-Synodinos, Danai Veltra, Afrodite Kampouraki, and George P. Chrousos. 2024. "A TMEM63A Nonsense Heterozygous Variant Linked to Infantile Transient Hypomyelinating Leukodystrophy Type 19?" Genes 15, no. 5: 525. https://doi.org/10.3390/genes15050525
APA StyleSiori, D., Vlachakis, D., Makrythanasis, P., Traeger-Synodinos, J., Veltra, D., Kampouraki, A., & Chrousos, G. P. (2024). A TMEM63A Nonsense Heterozygous Variant Linked to Infantile Transient Hypomyelinating Leukodystrophy Type 19? Genes, 15(5), 525. https://doi.org/10.3390/genes15050525