Epigenetic Control of the Genome—Lessons from Genomic Imprinting

{kind=link}
Abstract
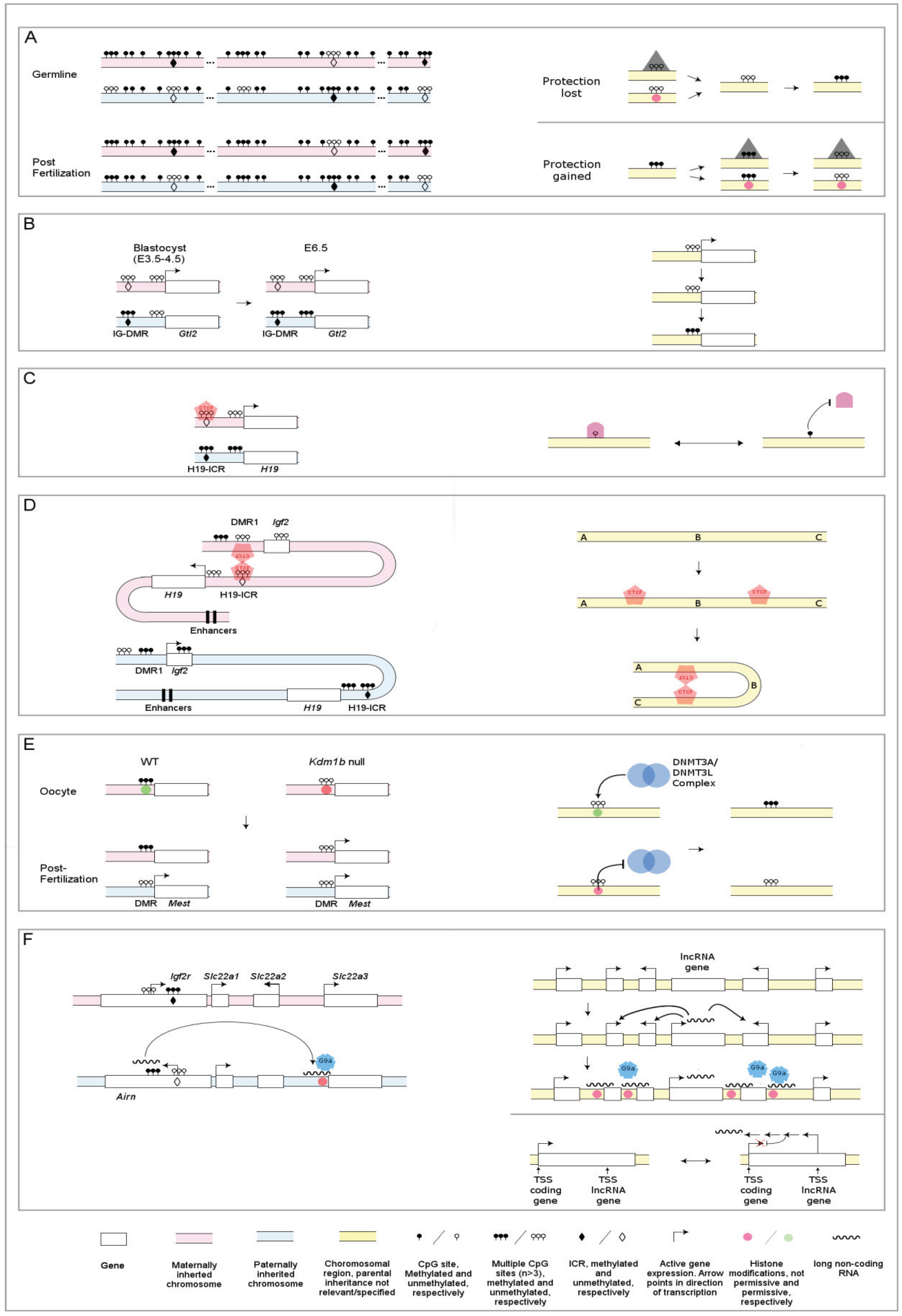
:1. A Primer on Epigenetics, DNA Methylation and Histone Modifications
2. Genomic Imprinting and Targeting DNA Methylation
3. DNA Methylation and Gene Repression—The Chicken or the Egg?
3.1. DNA Methylation Correlates with Repression
3.2. DNA Methylation as a Consequence of Transcriptional Silencing
4. How Does DNA Methylation Confer Effects on Gene Expression?
4.1. Proteins Attracted and Repelled
4.2. Regulation of CTCF Binding at the H19/Igf2 Imprinted Cluster; the Insulator Mechanism
5. Relationship between DNA Methylation and Histone Modifications

6. lncRNAs
6.1. lncRNAs, Definition, Characterization and Potential Functions
6.2. lncRNAs in the Epigenetic Control of Genome Function—Lessons from Imprinting
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity (Edinb). 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Kelsey, G. De novo DNA methylation: A germ cell perspective. Trends Genet. 2012, 28, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 2004, 429, 900–903. [Google Scholar] [PubMed]
- Gu, T.-P.; Guo, F.; Yang, H.; Wu, H.-P.; Xu, G.-F.; Liu, W.; Xie, Z.-G.; Shi, L.; He, X.; Jin, S.; et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Smith, A.C. Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011, 12, 565–575. [Google Scholar] [PubMed]
- Edwards, C.A.; Ferguson-Smith, A.C. Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 2007, 19, 281–289. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [PubMed]
- Surani, M.A.H.; Barton, S.C.; Norris, M.L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984, 308, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Smith, A.C.; Sasaki, H.; Cattanach, B.M.; Surani, M.A. Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature 1993, 362, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Stöger, R.; Kubicka, P.; Liu, C.-G.; Kafri, T.; Razin, A.; Cedar, H.; Barlow, D.P. Maternal-specific methylation of the imprinted mouse Igf2r locus identifies the expressed locus as carrying the imprinting signal. Cell 1993, 73, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Tomizawa, S.; Krueger, F.; Ruf, N.; Carli, N.; Segonds-Pichon, A.; Sato, S.; Hata, K.; Andrews, S.R.; Kelsey, G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 2011, 43, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, G.; Feil, R. New insights into establishment and maintenance of DNA methylation imprints in mammals. Phil. Trans. R. Soc. B 2013, 368. [Google Scholar] [CrossRef]
- Kobayashi, H.; Sakurai, T.; Imai, M.; Takahashi, N.; Fukuda, A.; Yayoi, O.; Sato, S.; Nakabayashi, K.; Hata, K.; Sotomaru, Y.; et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012, 8, e1002440. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Chan, M.M.; Mikkelsen, T.S.; Gu, H.; Gnirke, A.; Regev, A.; Meissner, A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 2012, 484, 339–344. [Google Scholar] [PubMed]
- Li, X.; Ito, M.; Zhou, F.; Youngson, N.; Zuo, X.; Leder, P.; Ferguson-Smith, A.C. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev. Cell 2008, 15, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Groudine, M.; Eisenman, R.; Weintraub, H. Chromatin structure of endogenous retroviral genes and activation by an inhibitor of DNA methylation. Nature 1981, 292, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Sutter, D.; Doerfler, W. Methylation of integrated adenovirus type 12 DNA sequences in transformed cells is inversely correlated with viral gene expression. Proc. Natl. Acad. Sci. USA 1980, 77, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Desrosiers, R.C.; Mulder, C.; Fleckenstein, B. Methylation of Herpesvirus saimiri DNA in lymphoid tumor cell lines. Proc. Natl. Acad. Sci. USA 1979, 76, 3839–3843. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C. Methylation of milk-borne and genetically transmitted mouse mammary tumor virus proviral DNA. Cell 1980, 19, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Guntaka, R.V.; Rao, P.Y.; Mitsialis, S.A.; Katz, R. Modification of avian sarcoma proviral DNA sequences in nonpermissive XC cells but not in permissive chicken cells. J. Virol. 1980, 34, 569–572. [Google Scholar] [PubMed]
- Van der Ploeg, L.H.T.; Flavell, R.A. DNA methylation in the human globin locus in erythroid and nonerythroid tissues. Cell 1980, 19, 947–958. [Google Scholar] [PubMed]
- McGhee, J.D.; Ginder, G.D. Specific DNA methylation sites in the vicinity of the chicken beta-globin genes. Nature 1979, 280, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Mandel, J.L.; Chambon, P. DNA methylation: Correlation with DNase I sensitivity of chicken ovalbumun and conalbumin chromatin. Nucleic Acids Res. 1979, 7, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Mandel, J.L.; Chambon, P. DNA methylation: Organ specific variations in the methylation pattern within and around ovalbumin and other chicken genes. Nucleic Acids Res. 1979, 7, 2081–2103. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P.; Taggart, M.H.; Smith, B.A. Methylated and unmethylated DNA compartments in the sea urchin genome. Cell 1979, 17, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.; Taggart, M.; Macleod, D. Loss of rDNA methylation accompanies the onset of ribosomal gene activity in early development of X. laevis. Cell 1981, 26, 381–390. [Google Scholar]
- Vardimon, L.; Kressmann, A.; Cedar, H.; Maechler, M.; Doerfler, W. Expression of a cloned adenovirus gene is inhibited by in vitro methylation. Proc. Natl. Acad. Sci. USA 1982, 79, 1073–1077. [Google Scholar] [PubMed]
- Stein, R.; Razin, A.; Cedar, H. In vitro methylation of the hamster adenine phosphoribosyltransferase gene inhibits its expression in mouse L cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3418–3422. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.-H.; LeProust, E.M.; Park, I.-H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; Antosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.-H.; Yu, J.; et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [PubMed]
- Rauch, T.A.; Wu, X.; Zhong, X.; Riggs, A.D.; Pfeifer, G.P. A human B cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 671–678. [Google Scholar] [PubMed]
- Trowbridge, J.J.; Orkin, S.H. DNA methylation in adult stem cells. Epigenetics 2010, 5, 189–193. [Google Scholar] [PubMed]
- Razin, A.; Cedar, H. DNA methylation and gene expression. Microbiol. Rev. 1991, 55, 451–458. [Google Scholar] [PubMed]
- Sato, S.; Yoshida, W.; Soejima, H.; Nakabayashi, K.; Hata, K. Methylation dynamics of IG-DMR and Gtl2-DMR during murine embryonic and placental development. Genomics 2011, 98, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, B.; Arnaudo, A.; Dymkowski, A.; Best, A.; Davis, T.L. Methylation at mouse Cdkn1c is acquired during postimplantation development and functions to maintain imprinted expression. Genomics 2004, 84, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Umlauf, D.; Goto, Y.; Cao, R.; Cerqueira, F.; Wagschal, A.; Zhang, Y.; Feil, R. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat. Genet. 2004, 36, 1296–1300. [Google Scholar] [PubMed]
- Sasaki, H.; Ferguson-Smith, A.C.; Shum, A.S.W.; Barton, S.C.; Surani, M.A. Temporal and spatial regulation of H19 imprinting in normal and uniparental mouse embryos. Development 1995, 121, 4195–4202. [Google Scholar] [PubMed]
- Lerchner, W.; Barlow, D.P. Paternal repression of the imprinted mouse Igf2r locus occurs during implantation and is stable in all tissues of the post-implantation mouse embryo. Mech. Dev. 1997, 61, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [PubMed]
- Meehan, R.R.; Lewis, J.D.; McKay, S.; Kleiner, E.L.; Bird, A.P. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 1989, 58, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Bird, A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [PubMed]
- Hendrich, B.; Tweedie, S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 2003, 19, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P.; Wolffe, A.P. Methylation-induced repression—Belts, braces, and chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Veenstra, G.J.C.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.-H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [PubMed]
- Ng, H.-H.; Zhang, Y.; Hendrich, B.; Johnson, C.A.; Turner, B.M.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D.; Bird, A. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 1999, 23, 58–61. [Google Scholar] [PubMed]
- Wade, P.A.; Gegonne, A.; Jones, P.L.; Ballestar, E.; Aubry, F.; Wolffe, A.P. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat. Genet. 1999, 23, 62–66. [Google Scholar] [PubMed]
- Zhang, Y.; Ng, H.-H.; Erdjument-Bromage, H.; Tempst, P.; Bird, A.; Reinberg, D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999, 13, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Stancheva, I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell 2004, 15, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Lawe, D.; Ziff, E.B. Association of Myn, the murine homolog of Max, with c-Myc stimulates methylation-sensitive DNA binding and Ras cotransformation. Cell 1991, 65, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.; Molloy, P.L. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988, 2, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Comb, M.; Goodman, H.M. CpG methylation inhibits proenkephalin gene expression and binding of the transcription factor AP-2. Nucleic Acids Res. 1990, 18, 3975–3982. [Google Scholar] [CrossRef] [PubMed]
- DeChiara, T.M.; Robertson, E.J.; Efstratiadis, A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell 1991, 64, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsen, J.L.; Duran, K.L.; Bartolomei, M.S. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 1998, 12, 3693–3702. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, K.D.; Duran, K.L.; Bartolomei, M.S. A 5' 2-kilobase-pair region of the imprinted mouse H19 gene exhibits exclusive paternal methylation throughout development. Mol. Cell. Biol. 1997, 17, 4322–4329. [Google Scholar] [PubMed]
- Szabó, P.E.; Tang, S.-H.E.; Rentsendorj, A.; Pfeifer, G.P.; Mann, J.R. Maternal-specific footprints at putative CTCF sites in the H19 imprinting control region give evidence for insulator function. Curr. Biol. 2000, 10, 607–610. [Google Scholar] [PubMed]
- Kanduri, C.; Pant, V.; Loukinov, D.; Pugacheva, E.; Qi, C.-F.; Wolffe, A.; Ohlsson, R.; Lobanenkov, V.V. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr. Biol. 2000, 10, 853–856. [Google Scholar] [CrossRef] [PubMed]
- Hark, A.T.; Schoenherr, C.J.; Katz, D.J.; Ingram, R.S.; Levorse, J.M.; Tilghman, S.M. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000, 405, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Webber, A.L.; Ingram, R.S.; Levorse, J.M.; Tilghman, S.M. Location of enhancers is essential for the imprinting of H19 and Igf2 genes. Nature 1998, 391, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Leighton, P.A.; Saam, J.R.; Ingram, R.S.; Stewart, C.L.; Tilghman, S.M. An enhancer deletion affects both H19 and Igf2 expression. Genes Dev. 1995, 9, 2079–2089. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [PubMed]
- Pant, V.; Kurukuti, S.; Pugacheva, E.; Shamsuddin, S.; Mariano, P.; Renkawitz, R.; Klenova, E.; Lobanenkov, V.; Ohlsson, R. Mutation of a single CTCF target site within the H19 imprinting control region leads to loss of Igf2 imprinting and complex patterns of de novo methylation upon maternal inheritance. Mol. Cell. Biol. 2004, 24, 3497–3504. [Google Scholar] [CrossRef] [PubMed]
- Yusufzai, T.M.; Tagami, H.; Nakatani, Y.; Felsenfeld, G. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol. Cell 2004, 13, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; Jeong, S.; Rong, Q.; Park, K.-Y.; Chung, J.H.; Pfeifer, K. Analysis of the H19ICR insulator. Mol. Cell. Biol. 2007, 27, 3499–3510. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hu, J.-F.; Qiu, X.; Ling, J.; Chen, H.; Wang, S.; Hou, A.; Vu, T.H.; Hoffman, A.R. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol. Cell. Biol. 2008, 28, 6473–6482. [Google Scholar] [CrossRef] [PubMed]
- Kurukuti, S.; Tiwari, V.K.; Tavoosidana, G.; Pugacheva, E.; Murrell, A.; Zhao, Z.; Lobanenkov, V.; Reik, W.; Ohlsson, R. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc. Natl. Acad. Sci. USA 2006, 103, 10684–10689. [Google Scholar] [CrossRef] [PubMed]
- Murrell, A.; Heeson, S.; Reik, W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat. Genet. 2004, 36, 889–893. [Google Scholar] [PubMed]
- Wendt, K.S.; Yoshida, K.; Itoh, T.; Bando, M.; Koch, B.; Schirghuber, E.; Tsutsumi, S.; Nagae, G.; Ishihara, K.; Mishiro, T.; et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008, 451, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Hon, G.C.; Hawkins, R.D.; Ren, B. Predictive chromatin signatures in the mammalian genome. Hum. Mol. Genet. 2009, 18, R195–R201. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Hon, G.; Wang, W.; Ren, B. Discovery and annotation of functional chromatin signatures in the human genome. PLoS Comput. Biol. 2009, 5, e1000566. [Google Scholar] [CrossRef] [PubMed]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, V.; O’Neill, L.; Sado, T.; Turner, B.; Ferguson-Smith, A. Relationship between DNA methylation, histone H4 acetylation and gene expression in the mouse imprinted Igf2-H19 domain. FEBS Lett. 2001, 488, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Pedone, P.V.; Pikaart, M.J.; Cerrato, F.; Vernucci, M.; Ungaro, P.; Bruni, C.B.; Riccio, A. Role of histone acetylation and DNA methylation in the maintenance of the imprinted expression of the H19 and Igf2 genes. FEBS Lett. 1999, 458, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, T. Mechanistic and functional links between histone methylation and DNA methylation. Prog. Mol. Biol. Transl. Sci. 2011, 101, 335–348. [Google Scholar]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [PubMed]
- Bourc’his, D.; Xu, G.-L.; Lin, C.-S.; Bollman, B.; Bestor, T.H. Dnmt3L and the establishment of maternal genomic imprints. Science 2001, 294, 2536–2539. [Google Scholar] [CrossRef] [PubMed]
- Bourc’his, D.; Bestor, T.H. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 2004, 431, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Jurkowska, R.Z.; Zhang, X.; Jeltsch, A.; Cheng, X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007, 449, 248–251. [Google Scholar] [PubMed]
- Margot, J.B.; Ehrenhofer-Murray, A.E.; Leonhardt, H. Interactions within the mammalian DNA methyltransferase family. BMC Mol. Biol. 2003, 4. [Google Scholar] [CrossRef] [Green Version]
- Suetake, I.; Shinozaki, F.; Miyagawa, J.; Takeshima, H.; Tajima, S. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem. 2004, 279, 27816–27823. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.T.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.-P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, D.N.; Su, H.; Hevi, S.; Gay, F.; Lei, H.; Bajko, J.; Xu, G.; Li, E.; Chen, T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009, 461, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, K.; Iwatani, M.; Suzuki, M.; Tachibana, M.; Shinkai, Y.; Tanaka, S.; Greally, J.M.; Yagi, S.; Hattori, N.; Shiota, K. Genome-wide and locus-specific DNA hypomethylation in G9a deficient mouse embryonic stem cells. Genes Cells 2007, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Matsumura, Y.; Fukuda, M.; Kimura, H.; Shinkai, Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 2008, 27, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.B.; Maksakova, I.A.; Mohn, F.; Leung, D.; Appanah, R.; Lee, S.; Yang, H.W.; Lam, L.L.; Mager, D.L.; Schübeler, D.; et al. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J. 2008, 27, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.H.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; Li, E.; Jenuwein, T.; Peters, A.H.F.M. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003, 13, 1192–1200. [Google Scholar] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Rauch, T.; Chen, Z.-X.; Szabó, P.E.; Riggs, A.D.; Pfeifer, G.P. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 2006, 281, 19489–19500. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.P.; Lindroth, A.M.; Cao, X.; Jacobsen, S.E. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 2002, 416, 556–560. [Google Scholar] [PubMed]
- Freitag, M.; Hickey, P.C.; Khlafallah, T.K.; Read, N.D.; Selker, E.U. HP1 is essential for DNA methylation in Neurospora. Mol. Cell 2004, 13, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.; Tachibana, M.; Guggiari, M.; Heard, E.; Shinkai, Y.; Wagstaff, J. Role of histone methyltransferase G9a in CpG methylation of the Prader-Willi syndrome imprinting center. J. Biol. Chem. 2003, 278, 14996–15000. [Google Scholar] [CrossRef] [PubMed]
- Wagschal, A.; Sutherland, H.G.; Woodfine, K.; Henckel, A.; Chebli, K.; Schulz, R.; Oakey, R.J.; Bickmore, W.A.; Feil, R. G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol. Cell. Biol. 2008, 28, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, F.; Mondal, T.; Kanduri, C. Epigenetics of imprinted long noncoding RNAs. Epigenetics 2009, 4, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Gerhardt, D.J.; Dinger, M.E.; Crawford, J.; Trapnell, C.; Jeddeloh, J.A.; Mattick, J.S.; Rinn, J.L. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 2012, 30, 99–104. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Latos, P.A.; Pauler, F.M.; Koerner, M.V; Senergin, H.B.; Hudson, Q.J.; Stocsits, R.R.; Allhoff, W.; Stricker, S.H.; Klement, R.M.; Warczok, K.E.; et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 2012, 338, 1469–1472. [Google Scholar]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R.; Frase, P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008, 322, 1717–1720. [Google Scholar] [CrossRef] [PubMed]
- Osato, N.; Suzuki, Y.; Ikeo, K.; Gojobori, T. Transcriptional interferences in cis natural antisense transcripts of humans and mice. Genetics 2007, 176, 1299–1306. [Google Scholar] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Howell, C.Y.; Bestor, T.H.; Ding, F.; Latham, K.E.; Mertineit, C.; Trasler, J.M.; Chaillet, J.R. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 2001, 104, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Mitsuya, K.; Umlauf, D.; Smith, P.; Dean, W.; Walter, J.; Higgins, M.; Feil, R.; Reik, W. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat. Genet. 2004, 36, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-DiNardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef]
- Seidl, C.I.M.; Stricker, S.H.; Barlow, D.P. The imprinted Air ncRNA is an atypical RNAPII transcript that evades splicing and escapes nuclear export. EMBO J. 2006, 25, 3565–3575. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-Y.; Fitzpatrick, G.V.; Higgins, M.J. Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. EMBO J. 2008, 27, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Mancini-DiNardo, D.; Steele, S.J.S.; Levorse, J.M.; Ingram, R.S.; Tilghman, S.M. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006, 20, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002, 415, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Zwart, R.; Sleutels, F.; Wutz, A.; Schinkel, A.H.; Barlow, D.P. Bidirectional action of the Igf2r imprint control element on upstream and downstream imprinted genes. Genes Dev. 2001, 15, 2361–2366. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Morales, R.D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, S.; Bonasio, R.; Saldaña-Meyer, R.; Yoshida, T.; Son, J.; Nishino, K.; Umezawa, A.; Reinberg, D. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol. Cell 2014, 53, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Arai, Y.; Umehara, H.; Masuhara, M.; Kimura, T.; Taniguchi, H.; Sekimoto, T.; Ikawa, M.; Yoneda, Y.; Okabe, M.; et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat. Cell Biol. 2007, 9, 64–71. [Google Scholar] [PubMed]
- Ferrón, S.R.; Charalambous, M.; Radford, E.; McEwen, K.; Wildner, H.; Hind, E.; Morante-Redolat, J.M.; Laborda, J.; Guillemot, F.; Bauer, S.R.; et al. Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature 2011, 475, 381–385. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Adalsteinsson, B.T.; Ferguson-Smith, A.C. Epigenetic Control of the Genome—Lessons from Genomic Imprinting. Genes 2014, 5, 635-655. https://doi.org/10.3390/genes5030635
Adalsteinsson BT, Ferguson-Smith AC. Epigenetic Control of the Genome—Lessons from Genomic Imprinting. Genes. 2014; 5(3):635-655. https://doi.org/10.3390/genes5030635
Chicago/Turabian StyleAdalsteinsson, Bjorn T., and Anne C. Ferguson-Smith. 2014. "Epigenetic Control of the Genome—Lessons from Genomic Imprinting" Genes 5, no. 3: 635-655. https://doi.org/10.3390/genes5030635
APA StyleAdalsteinsson, B. T., & Ferguson-Smith, A. C. (2014). Epigenetic Control of the Genome—Lessons from Genomic Imprinting. Genes, 5(3), 635-655. https://doi.org/10.3390/genes5030635