
Yeast Phenomics: An Experimental Approach for Modeling Gene Interaction Networks that Buffer Disease

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
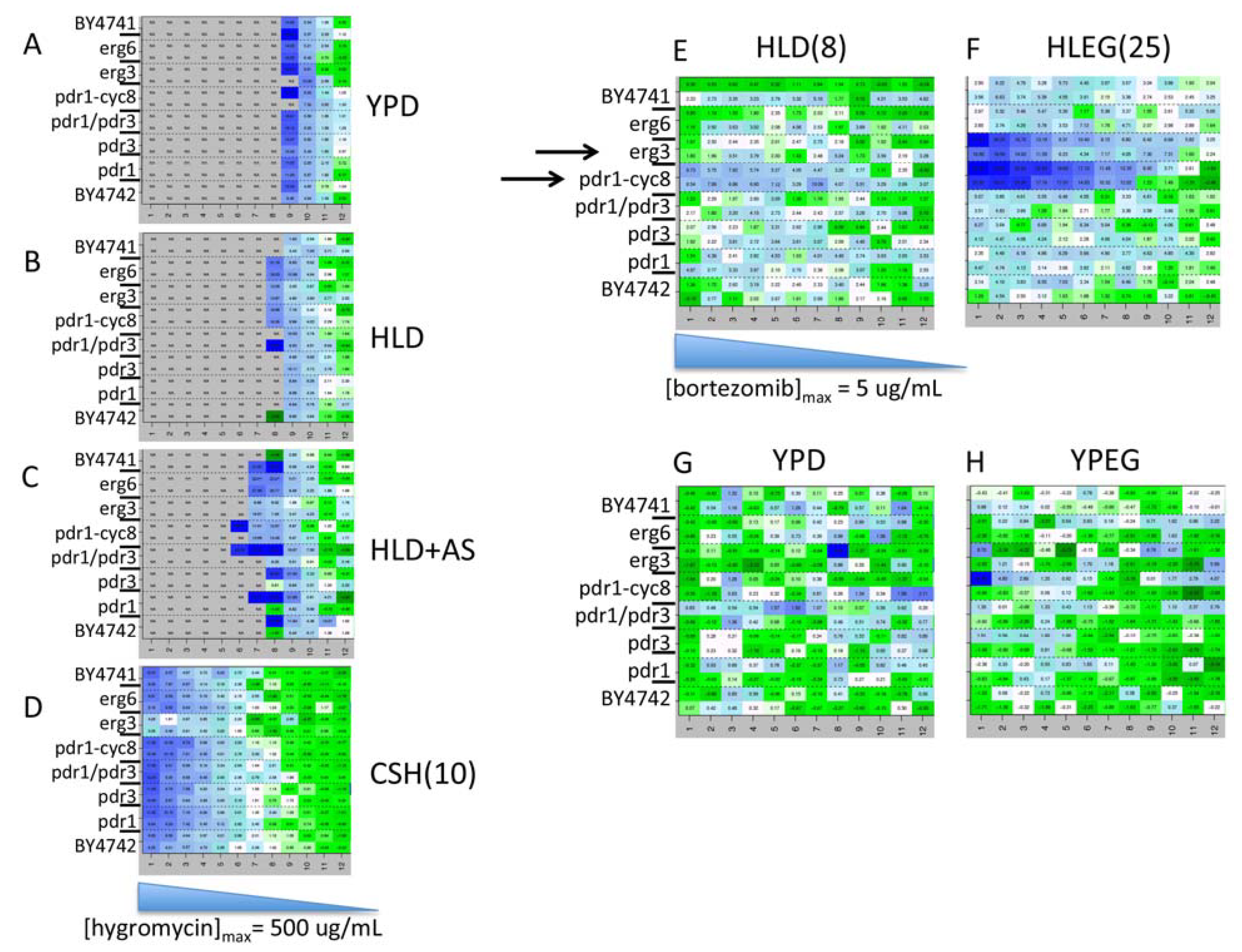{kind=link}
{kind=link}
1. Introduction and Perspective
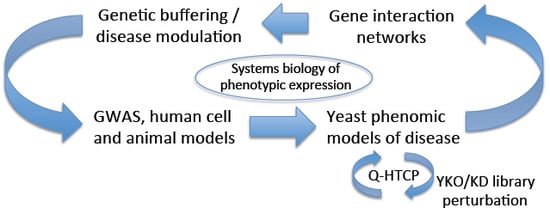
1.1. Buffering of Phenotypes: Yeast Phenomic Analysis Reveals Gene Interaction Networks Responsible for Phenotypic Variability
1.2. The Need for Quantitative Phenotyping to Experimentally Derive Buffering Networks
1.3. Are Gene Interaction Networks That Buffer Human Disease Evolutionarily Conserved?
1.4. Experimental Resources and Technology for Yeast Phenomic Analysis
1.5. Examples of Yeast Phenomic Modeling of Disease in Our Laboratory
1.6. Development of a Human-Like (HL) Media for Yeast Phenomic Studies
2. Methods
2.1. Yeast Media and Strains
2.2. Quantitative High Throughput Cell Array Phenotyping (Q-HTCP)
2.3. Quantification of Gene Interaction
2.4. Recursive Expectation Maximization Clustering (REMc)
3. Results
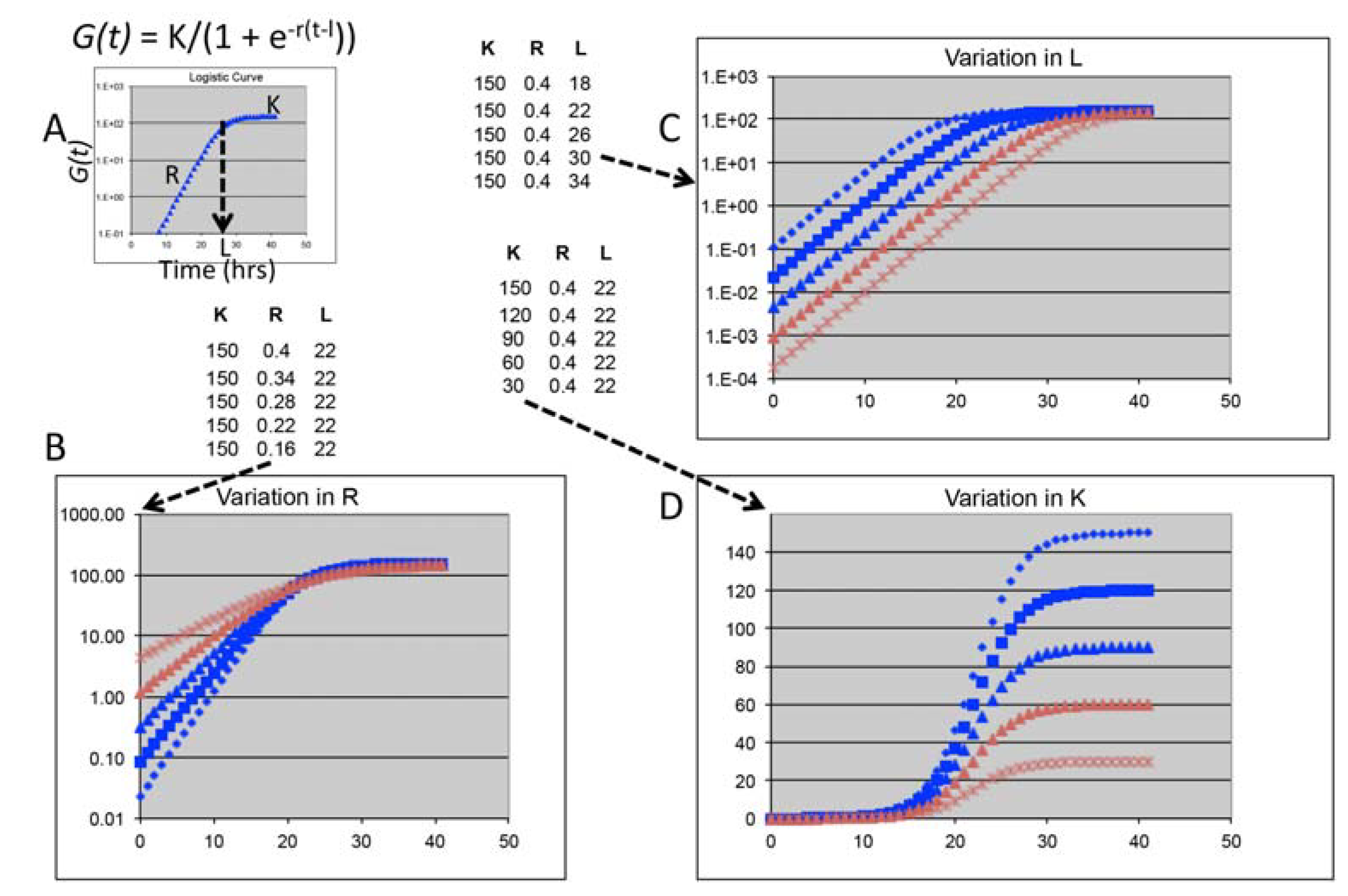
3.1. Using Growth Curve Parameters as High-Resolution, Quantitative Phenotypes

3.2. A Human-Like Yeast Media to Increase Positive Predictive Value of Yeast Phenomic Models

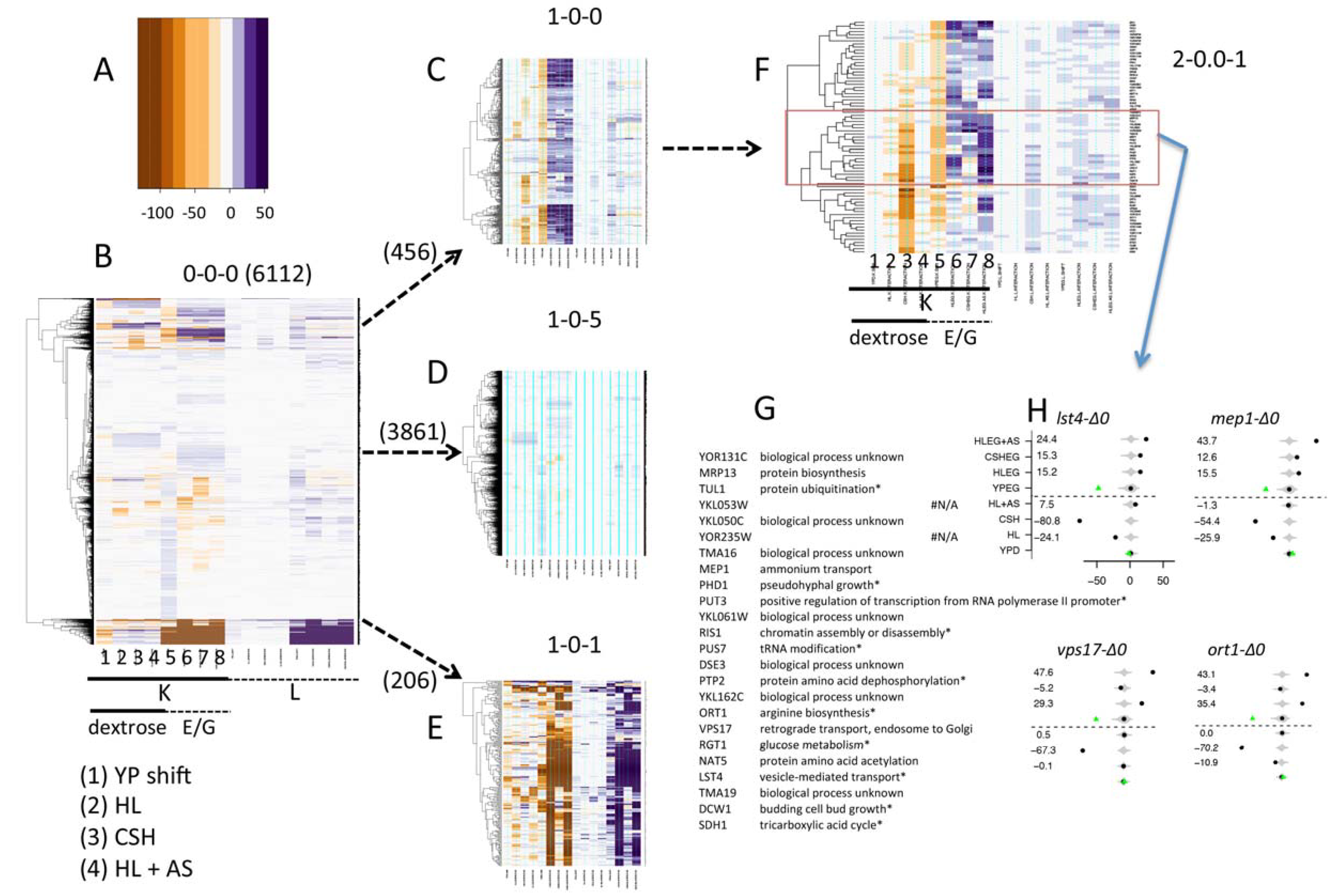
3.3. Phenomic Analysis Reveals Clusters of Gene X Media Interaction

3.4. Resolving Drug-Media Interaction by Q-HTCP across Drug-Gradients

4. Discussion

5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lupski, J.R.; Belmont, J.W.; Boerwinkle, E.; Gibbs, R.A. Clan genomics and the complex architecture of human disease. Cell 2011, 147, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Katsanis, N. Beyond mendel: An evolving view of human genetic disease transmission. Nat. Rev. Genet. 2002, 3, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Hartman, J.L., 4th; Garvik, B.; Hartwell, L. Principles for the buffering of genetic variation. Science 2001, 291, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Costanzo, M.; Baryshnikova, A.; Andrews, B.; Boone, C. Systematic mapping of genetic interaction networks. Annu. Rev. Genet. 2009, 43, 601–625. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Baryshnikova, A.; Myers, C.L.; Andrews, B.; Boone, C. Charting the genetic interaction map of a cell. Curr. Opin. Biotechnol. 2011, 22, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Lehner, B.; Vavouri, T.; Shin, J.; Fraser, A.G.; Marcotte, E.M. Predicting genetic modifier loci using functional gene networks. Genome Res. 2010, 20, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- McGary, K.L.; Park, T.J.; Woods, J.O.; Cha, H.J.; Wallingford, J.B.; Marcotte, E.M. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc. Natl. Acad. Sci. USA 2010, 107, 6544–6549. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.O.; Singh-Blom, U.M.; Laurent, J.M.; McGary, K.L.; Marcotte, E.M. Prediction of gene-phenotype associations in humans, mice, and plants using phenologs. BMC Bioinform. 2013. [Google Scholar] [CrossRef]
- Hwang, S.; Kim, E.; Yang, S.; Marcotte, E.M.; Lee, I. Morphin: A web tool for human disease research by projecting model organism biology onto a human integrated gene network. Nucleic Acids Res. 2014, 42, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Louie, R.J.; Guo, J.; Rodgers, J.W.; White, R.; Shah, N.; Pagant, S.; Kim, P.; Livstone, M.; Dolinski, K.; McKinney, B.A.; et al. A yeast phenomic model for the gene interaction network modulating cftr-∆f508 protein biogenesis. Genome Med. 2012. [Google Scholar] [CrossRef]
- Ratnakumar, S.; Hesketh, A.; Gkargkas, K.; Wilson, M.; Rash, B.M.; Hayes, A.; Tunnacliffe, A.; Oliver, S.G. Phenomic and transcriptomic analyses reveal that autophagy plays a major role in desiccation tolerance in Saccharomyces cerevisiae. Mol. Biosyst. 2011, 7, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tian, D.; McKinney, B.A.; Hartman, J.L., 4th. Recursive expectation-maximization clustering: A method for identifying buffering mechanisms composed of phenomic modules. Chaos 2010. [Google Scholar] [CrossRef]
- Kahraman, A.; Avramov, A.; Nashev, L.G.; Popov, D.; Ternes, R.; Pohlenz, H.D.; Weiss, B. PhenomicDB: A multi-species genotype/phenotype database for comparative phenomics. Bioinformatics 2005, 21, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Warringer, J.; Ericson, E.; Fernandez, L.; Nerman, O.; Blomberg, A. High-resolution yeast phenomics resolves different physiological features in the saline response. Proc. Natl. Acad. Sci. USA 2003, 100, 15724–15729. [Google Scholar] [CrossRef] [PubMed]
- Parsons, A.B.; Brost, R.L.; Ding, H.; Li, Z.; Zhang, C.; Sheikh, B.; Brown, G.W.; Kane, P.M.; Hughes, T.R.; Boone, C. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat. Biotechnol. 2004, 22, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Hartman, J.L., 4th; Tippery, N.P. Systematic quantification of gene interactions by phenotypic array analysis. Genome Biol. 2004. [Google Scholar] [CrossRef] [Green Version]
- Hillenmeyer, M.E.; Fung, E.; Wildenhain, J.; Pierce, S.E.; Hoon, S.; Lee, W.; Proctor, M.; Onge, R.P., St.; Tyers, M.; Koller, D.; et al. The chemical genomic portrait of yeast: Uncovering a phenotype for all genes. Science 2008, 320, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Heinicke, S.; Boucher, L.; Winter, A.; Stark, C.; Nixon, J.; Ramage, L.; Kolas, N.; O’Donnell, L.; et al. The biogrid interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D816–D823. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.H.; Boone, C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol. Biol. 2006, 313, 171–192. [Google Scholar] [PubMed]
- Singh, I.; Pass, R.; Togay, S.O.; Rodgers, J.W.; Hartman, J.L., 4th. Stringent mating-type-regulated auxotrophy increases the accuracy of systematic genetic interaction screens with Saccharomyces cerevisiae mutant arrays. Genetics 2009, 181, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Baryshnikova, A.; Bellay, J.; Kim, Y.; Spear, E.D.; Sevier, C.S.; Ding, H.; Koh, J.L.; Toufighi, K.; Mostafavi, S.; et al. The genetic landscape of a cell. Science 2010, 327, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Schuldiner, M.; Collins, S.R.; Thompson, N.J.; Denic, V.; Bhamidipati, A.; Punna, T.; Ihmels, J.; Andrews, B.; Boone, C.; Greenblatt, J.F.; et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 2005, 123, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Ihmels, J.; Collins, S.R.; Schuldiner, M.; Krogan, N.J.; Weissman, J.S. Backup without redundancy: Genetic interactions reveal the cost of duplicate gene loss. Mol. Syst. Biol. 2007. [Google Scholar] [CrossRef]
- Jonikas, M.C.; Collins, S.R.; Denic, V.; Oh, E.; Quan, E.M.; Schmid, V.; Weibezahn, J.; Schwappach, B.; Walter, P.; Weissman, J.S.; et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 2009, 323, 1693–1697. [Google Scholar] [CrossRef] [PubMed]
- Boone, C. Yeast systems biology: Our best shot at modeling a cell. Genetics 2014, 198, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Warringer, J.; Blomberg, A. Automated screening in environmental arrays allows analysis of quantitative phenotypic profiles in Saccharomyces cerevisiae. Yeast 2003, 20, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.B.; Ehrenreich, I.M. Genetic interactions involving five or more genes contribute to a complex trait in yeast. PLOS Genet. 2014. [Google Scholar] [CrossRef]
- Dixon, S.J.; Fedyshyn, Y.; Koh, J.L.; Prasad, T.S.; Chahwan, C.; Chua, G.; Toufighi, K.; Baryshnikova, A.; Hayles, J.; Hoe, K.L.; et al. Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proc. Natl. Acad. Sci. USA 2008, 105, 16653–16658. [Google Scholar] [CrossRef] [PubMed]
- Roguev, A.; Bandyopadhyay, S.; Zofall, M.; Zhang, K.; Fischer, T.; Collins, S.R.; Qu, H.; Shales, M.; Park, H.O.; Hayles, J.; et al. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science 2008, 322, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, A. Measuring growth rate in high-throughput growth phenotyping. Curr. Opin. Biotechnol. 2011, 22, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Veronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; Andre, B.; et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.R.; Schuldiner, M.; Krogan, N.J.; Weissman, J.S. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 2006. [Google Scholar] [CrossRef]
- Baryshnikova, A.; Costanzo, M.; Kim, Y.; Ding, H.; Koh, J.; Toufighi, K.; Youn, J.Y.; Ou, J.; San Luis, B.J.; Bandyopadhyay, S.; et al. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat. Methods 2010, 7, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Liti, G.; Carter, D.M.; Moses, A.M.; Warringer, J.; Parts, L.; James, S.A.; Davey, R.P.; Roberts, I.N.; Burt, A.; Koufopanou, V.; et al. Population genomics of domestic and wild yeasts. Nature 2009, 458, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Onge, R.P., St.; Mani, R.; Oh, J.; Proctor, M.; Fung, E.; Davis, R.W.; Nislow, C.; Roth, F.P.; Giaever, G. Systematic pathway analysis using high-resolution fitness profiling of combinatorial gene deletions. Nat. Genet. 2007, 39, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.A.; Laws, R.J.; Wardman, B.; Zhao, L.P.; Hartman, J.L., 4th. Accurate, precise modeling of cell proliferation kinetics from time-lapse imaging and automated image analysis of agar yeast culture arrays. BMC Syst. Biol. 2007. [Google Scholar] [CrossRef]
- Schekman, R. Lasker basic medical research award. Sec mutants and the secretory apparatus. Nat. Med. 2002, 8, 1055–1058. [Google Scholar] [CrossRef]
- Hartwell, L.H. Nobel lecture. Yeast and cancer. Biosci. Rep. 2002, 22, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.; Outeiro, T.F.; DeVit, M.J.; Lindquist, S.L.; Muchowski, P.J. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science 2003, 302, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Caraveo, G.; Lindquist, S. Alpha-synuclein: Membrane interactions and toxicity in parkinson’s disease. Annu. Rev. Cell Dev. Biol. 2010, 26, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional links between abeta toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Armakola, M.; Higgins, M.J.; Figley, M.D.; Barmada, S.J.; Scarborough, E.A.; Diaz, Z.; Fang, X.; Shorter, J.; Krogan, N.J.; Finkbeiner, S.; et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat. Genet. 2012, 44, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The parkinson’s disease protein alpha-synuclein disrupts cellular rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.S.; McCaffery, J.M.; Lindquist, S.; Gitler, A.D. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar] [CrossRef] [PubMed]
- Couplan, E.; Aiyar, R.S.; Kucharczyk, R.; Kabala, A.; Ezkurdia, N.; Gagneur, J.; Onge, R.P., St.; Salin, B.; Soubigou, F.; le Cann, M.; et al. A yeast-based assay identifies drugs active against human mitochondrial disorders. Proc. Natl. Acad. Sci. USA 2011, 108, 11989–11994. [Google Scholar] [CrossRef] [PubMed]
- Vilaca, R.; Silva, E.; Nadais, A.; Teixeira, V.; Matmati, N.; Gaifem, J.; Hannun, Y.A.; Sa Miranda, M.C.; Costa, V. Sphingolipid signalling mediates mitochondrial dysfunctions and reduced chronological lifespan in the yeast model of niemann-pick type c1. Mol. Microbiol. 2014, 91, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Singh, K.K. Global genetic determinants of mitochondrial DNA copy number. PLOS ONE 2014, 9, e105242. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Dallabona, C.; Ferrero, I.; Frontali, L.; Bolotin-Fukuhara, M. Mitochondrial diseases and the role of the yeast models. FEMS Yeast Res. 2010, 10, 1006–1022. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Lehmann, R. Modeling human disease. Science 2012. [Google Scholar] [CrossRef]
- Mani, R.; Onge, R.P., St.; Hartman, J.L., 4th; Giaever, G.; Roth, F.P. Defining genetic interaction. Proc. Natl. Acad. Sci. USA 2008, 105, 3461–3466. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.; Guo, J.; Hartman, J.L., 4th. Phenomic assessment of genetic buffering by kinetic analysis of cell arrays. Methods Mol. Biol. 2014, 1205, 187–208. [Google Scholar] [PubMed]
- Carlborg, O.; Haley, C.S. Epistasis: Too often neglected in complex trait studies? Nat. Rev. Genet. 2004, 5, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.H.; Williams, S.M. Traversing the conceptual divide between biological and statistical epistasis: Systems biology and a more modern synthesis. Bioessays 2005, 27, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Leitch, C.C.; Ansley, S.J.; May-Simera, H.; Lawson, S.; Lewis, R.A.; Beales, P.L.; Dietz, H.C.; Fisher, S.; Katsanis, N. Dissection of epistasis in oligogenic bardet-biedl syndrome. Nature 2006, 439, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.C. Epistasis—The essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 2008, 9, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Cowper-Sal lari, R.; Cole, M.D.; Karagas, M.R.; Lupien, M.; Moore, J.H. Layers of epistasis: Genome-wide regulatory networks and network approaches to genome-wide association studies. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.S.; Kemena, C.; Vlasov, P.K.; Notredame, C.; Kondrashov, F.A. Epistasis as the primary factor in molecular evolution. Nature 2012, 490, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Drumm, M. The influence of genetics on cystic fibrosis phenotypes. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef]
- Hartman, J.L., 4th. Buffering of deoxyribonucleotide pool homeostasis by threonine metabolism. Proc. Natl. Acad. Sci. USA 2007, 104, 11700–11705. [Google Scholar] [CrossRef] [PubMed]
- Reichard, P. Ribonucleotide reductases: The evolution of allosteric regulation. Arch. Biochem. Biophys. 2002, 397, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.; Fernandes, P.A.; Ramos, M.J. Mechanism for ribonucleotide reductase inactivation by the anticancer drug gemcitabine. J. Comput. Chem. 2004, 25, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.; Pereira, S.; Fernandes, P.A.; Ramos, M.J. Overview of ribonucleotide reductase inhibitors: An appealing target in anti-tumour therapy. Curr. Med. Chem. 2005, 12, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Thelander, L. Ribonucleotide reductase and mitochondrial DNA synthesis. Nat. Genet. 2007, 39, 703–704. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Alexander, P.; Wu, L.; Hammer, R.; Cleaver, O.; McKnight, S.L. Dependence of mouse embryonic stem cells on threonine catabolism. Science 2009, 325, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.M.; Han, H.J. l-Threonine regulates G1/S phase transition of mouse embryonic stem cells via Pi3k/Akt, MAPKs, and mTORC pathways. J. Biol. Chem. 2011, 286, 23667–23678. [Google Scholar] [CrossRef] [PubMed]
- Shyh-Chang, N.; Locasale, J.W.; Lyssiotis, C.A.; Zheng, Y.; Teo, R.Y.; Ratanasirintrawoot, S.; Zhang, J.; Onder, T.; Unternaehrer, J.J.; Zhu, H.; et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 2013, 339, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Murakami, C.; Kaeberlein, M. Quantifying yeast chronological life span by outgrowth of aged cells. J. Vis. Exp. 2009. [Google Scholar] [CrossRef]
- Hess, D.C.; Lu, W.; Rabinowitz, J.D.; Botstein, D. Ammonium toxicity and potassium limitation in yeast. PLOS Biol. 2006. [Google Scholar] [CrossRef]
- Burke, D.; Dawson, D.; Stearns, T. Methods in Yeast Genetics; CSHL Press: Plainview, NY, USA, 2000. [Google Scholar]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. Go: Termfinder—Open source software for accessing gene ontology information and finding significantly enriched gene ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M. Lessons on longevity from budding yeast. Nature 2010, 464, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Sutphin, G.L.; Olsen, B.A.; Kennedy, B.K.; Kaeberlein, M. Genome-wide analysis of yeast aging. Subcell. Biochem. 2012, 57, 251–289. [Google Scholar] [PubMed]
- Roberg, K.J.; Bickel, S.; Rowley, N.; Kaiser, C.A. Control of amino acid permease sorting in the late secretory pathway of saccharomyces cerevisiae by SEC13, LST4, LST7 and LST8. Genetics 1997, 147, 1569–1584. [Google Scholar] [PubMed]
- Goldstein, A.L.; McCusker, J.H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 1999, 15, 1541–1553. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, A.; Nitiss, K.C.; Neale, G.; Nitiss, J.L. Enhancing drug accumulation in saccharomyces cerevisiae by repression of pleiotropic drug resistance genes with chimeric transcription repressors. Mol. Pharmacol. 2008, 74, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.A.; Lightcap, E.S.; Sadis, S.; Thoroddsen, V.; Bulawa, C.E.; Blackman, R.K. Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by ps-341. Proc. Natl. Acad. Sci. USA 2002, 99, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.A.; Kiser, G.L.; Hartwell, L.H. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994, 8, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Paulovich, A.G.; Hartwell, L.H. A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 1995, 82, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D. Why we need more basic biology research, not less. Mol. Biol. Cell 2012, 23, 4160–4161. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Clark, A.G.; Mootha, V.K. Distilling pathophysiology from complex disease genetics. Cell 2013, 155, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; Fink, G.R. Yeast: An experimental organism for 21st century biology. Genetics 2011, 189, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Dolinski, K.; Botstein, D. Orthology and functional conservation in eukaryotes. Annu. Rev. Genet. 2007, 41, 465–507. [Google Scholar] [CrossRef] [PubMed]
- Califano, A.; Butte, A.J.; Friend, S.; Ideker, T.; Schadt, E. Leveraging models of cell regulation and gwas data in integrative network-based association studies. Nat. Genet. 2012, 44, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Friend, S.H.; Schadt, E.E. Translational genomics. Clues from the resilient. Science 2014, 344, 970–972. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartman, J.L., IV; Stisher, C.; Outlaw, D.A.; Guo, J.; Shah, N.A.; Tian, D.; Santos, S.M.; Rodgers, J.W.; White, R.A. Yeast Phenomics: An Experimental Approach for Modeling Gene Interaction Networks that Buffer Disease. Genes 2015, 6, 24-45. https://doi.org/10.3390/genes6010024
Hartman JL IV, Stisher C, Outlaw DA, Guo J, Shah NA, Tian D, Santos SM, Rodgers JW, White RA. Yeast Phenomics: An Experimental Approach for Modeling Gene Interaction Networks that Buffer Disease. Genes. 2015; 6(1):24-45. https://doi.org/10.3390/genes6010024
Chicago/Turabian StyleHartman, John L., IV, Chandler Stisher, Darryl A. Outlaw, Jingyu Guo, Najaf A. Shah, Dehua Tian, Sean M. Santos, John W. Rodgers, and Richard A. White. 2015. "Yeast Phenomics: An Experimental Approach for Modeling Gene Interaction Networks that Buffer Disease" Genes 6, no. 1: 24-45. https://doi.org/10.3390/genes6010024
APA StyleHartman, J. L., IV, Stisher, C., Outlaw, D. A., Guo, J., Shah, N. A., Tian, D., Santos, S. M., Rodgers, J. W., & White, R. A. (2015). Yeast Phenomics: An Experimental Approach for Modeling Gene Interaction Networks that Buffer Disease. Genes, 6(1), 24-45. https://doi.org/10.3390/genes6010024