
Applicability of Next Generation Sequencing Technology in Microsatellite Instability Testing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Samples

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumours | MSI markers tested | |
|---|---|---|
| Multiplex PCR | NGS | |
| Series 1 * | BAT25 †, BAT26, CAT25, NR21, NR22, NR24, D5S346, D2S123, D17S250 | BAT25, BAT26, BAT34c4, D18S55, D5S346 |
| Series 2 ‡ | BAT25, BAT26, NR21, NR24, MONO27 | BAT25, BAT26 |
2.2. MSI Loci (Amplicons) Target Resequencing with MiSeq
| MSI loci | Position (chromosome) | Coordinates | Length (base pair) | Forward sequence | Reverse sequence |
|---|---|---|---|---|---|
| BAT25 | 4q12 | 55598151-55598274 | 123 | 5'-TCGCCTCCAAGAATGTAAGT-3' | 5'-TCTGCATTTTAACTATGGCTC-3' |
| BAT26 | 2p | 47641487-47641608 | 121 | 5'-TGACTACTTTTGACTTCAGCC-3' | 5'-AACCATTCAACATTTTTAACCC-3' |
| BAT34c4 | 17p13.1 | 7572124-7572254 | 130 | 5'-ACCCTGGAGGATTTCATCTC-3' | 5'-AACAAAGCGAGACCCAGTCT-3' |
| D18S55 | 18q22.1 | 61873501-61873648 | 147 | 5'-GGGAAGTCAAATGCAAATC-3' | 5'-AGCTTCTGAGTAATCTTATGCTGTG-3' |
| D5S346 | 5q22.2 | 112213624-112213748 | 124 | 5'-ACTCACTCTAGTGATAAATCGGG-3' | 5'-AGCAGATAAGACAGTATTACTAGTT-3' |
2.3. Sequencing
2.4. Data Analysis
2.4.1. Amplivar and SeqPrep for Processing NGS Data
2.4.2. Quantifying Amplicons
2.4.3. Defining Individual Locus Stability

2.4.4. Defining the Overall MSI Status for Each Tumour
3. Results
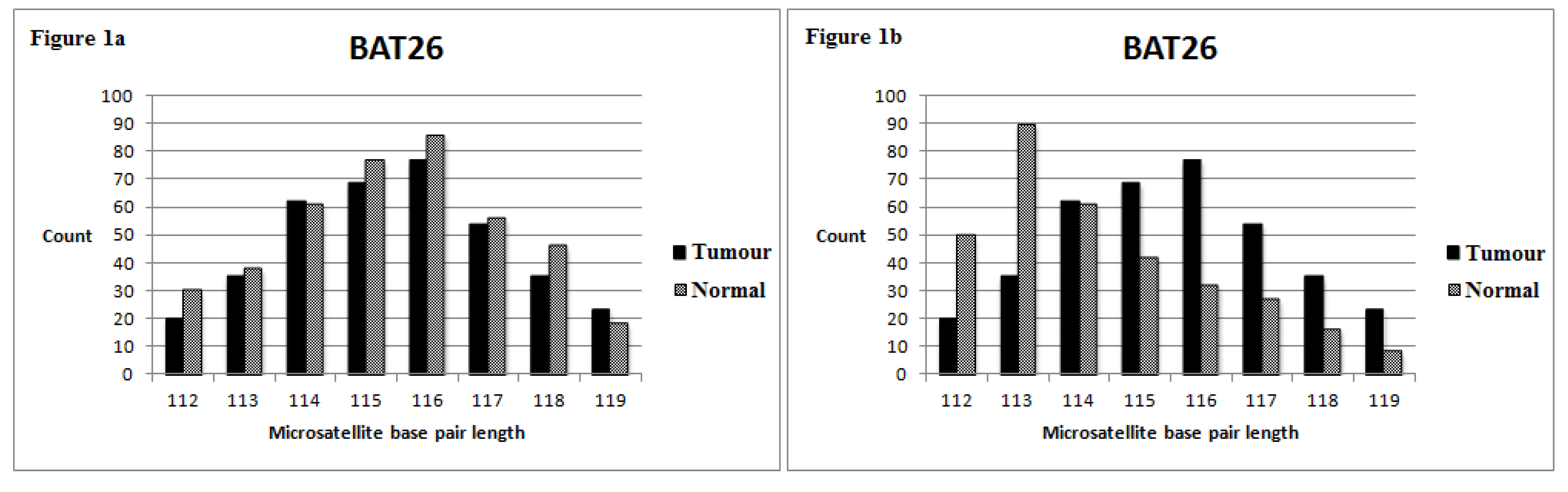
3.1. MiSeq Sequencing Profile
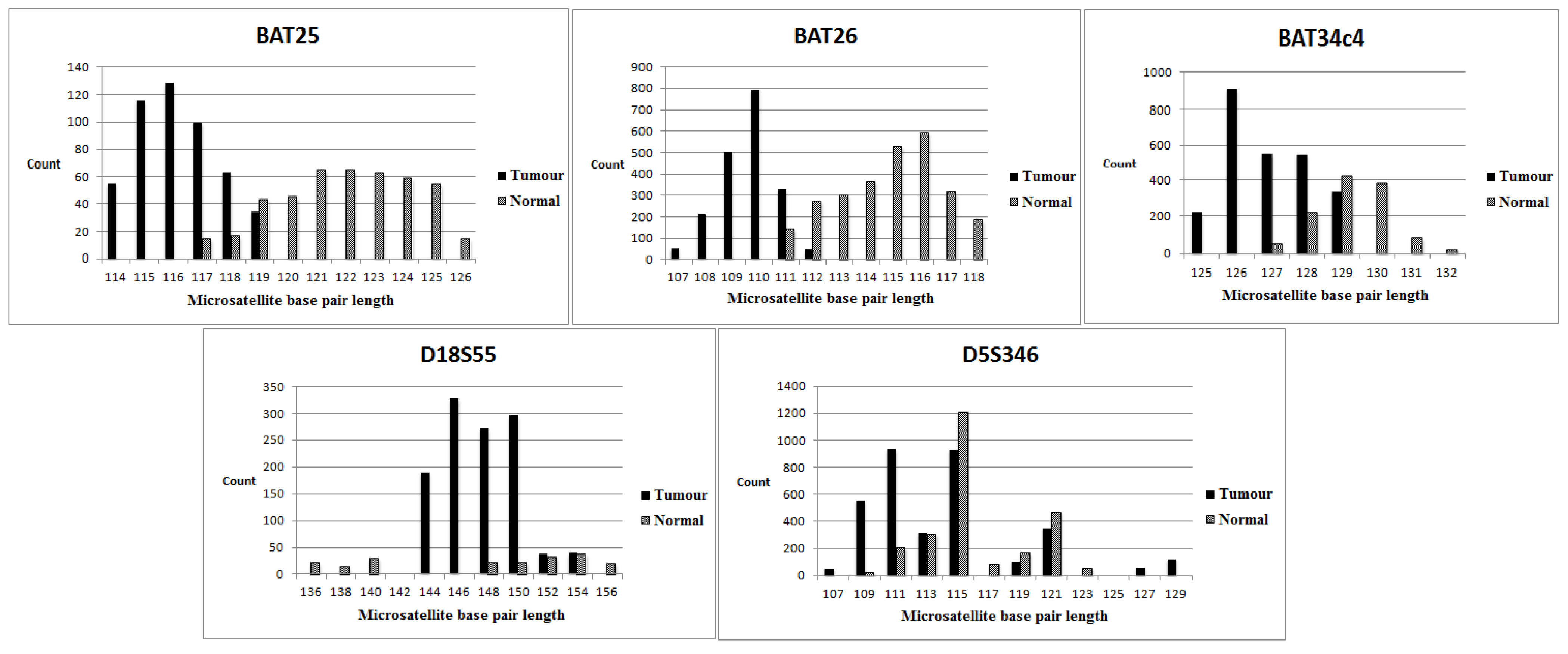
3.2. MSI Status of Series 1 (with Normal Tissue Control)
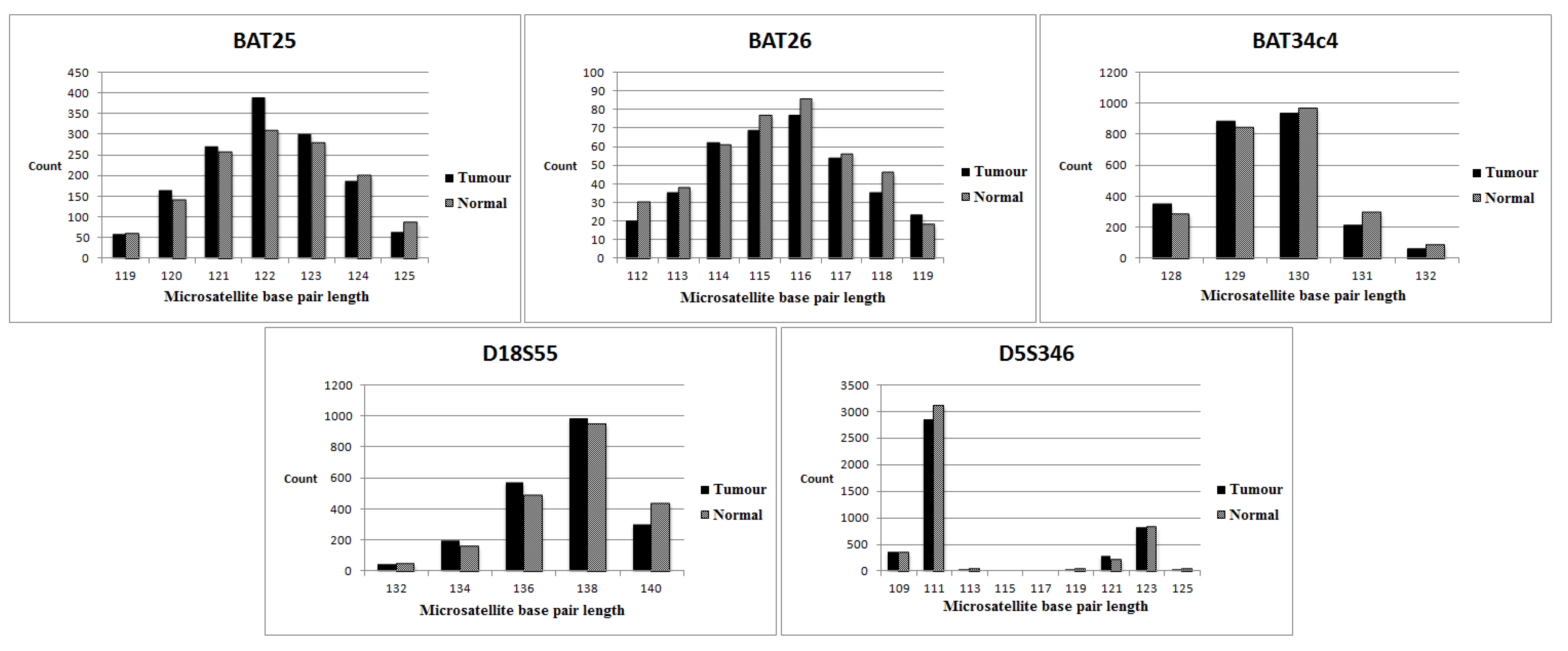
3.3. MSI Status of Series 2 (No Normal Tissue Control)
3.4. Evaluation of Individual Markers (NGS versus the Multiplex PCR-Based Method)
| Cases | BAT25 (M) (NGS/ Multiplex PCR) | BAT26 (M) (NGS/ Multiplex PCR) | D5S346 (D) (NGS/ Multiplex PCR) | BAT34c4 (M) (NGS only) | D18S55 (D) (NGS only) | MSI status (NGS) | MSI status (Multiplex PCR) |
|---|---|---|---|---|---|---|---|
| 1 | −/− | −/− | −/− | − | − | Stable | Stable |
| 2 | −/− | −/− | −/− | − | − | Stable | Stable |
| 3 | +/+ | +/+ | +/+ | + | − | High | High |
| 4 | +/+ | +/+ | +/+ | + | + | High | High |
| 5 | −/− | −/− | −/− | − | − | Stable | Stable |
| 6 | +/+ | +/+ | −/− | − | + | High | High |
| 7 | −/− | −/− | −/− | − | − | Stable | Stable |
| 8 | +/+ | +/+ | +/+ | + | + | High | High |
| 9 | +/+ | +/+ | −/− | − | − | High | High |
| 10 | +/+ | +/+ | −/− | + | + | High | High |
| 11 | −/− | −/− | −/− | − | − | Stable | Stable |
| 12 | −/− | −/− | −/− | − | − | Stable | Stable |
| 13 | −/− | −/− | −/− | − | − | Stable | Stable |
| 14 | −/− | −/− | −/− | − | − | Stable | Stable |
| 15 | +/+ | +/+ | −/− | + | + | High | High |
| 16 | −/− | −/− | −/− | − | − | Stable | Stable |
| 17 | −/− | −/− | −/− | − | − | Stable | Stable |
| 18 | +/+ | +/+ | −/+ FN | + | − | High | High |
| 19 | +/+ | +/+ | +/+ | + | + | High | High |
| 20 | −/− | −/− | −/− | − | − | Stable | Stable |
| 21 | −/− | −/− | −/− | − | + | Low | Stable |
| 22 | −/− | −/− | −/− | − | − | Stable | Stable |
| 23 | −/− | −/− | −/− | − | − | Stable | Stable |
| 24 | −/− | −/− | −/− | − | − | Stable | Stable |
| 25 | −/− | −/− | −/− | − | − | Stable | Stable |
| 26 | +/+ | +/+ | +/+ | − | + | High | High |
| 27 | +/+ | +/+ | −/− | + | + | High | High |
| 28 | −/− | −/− | −/− | − | − | Stable | Stable |
| 29 | +/+ | +/+ | −/− | + | − | High | High |
| 30 | −/− | −/− | −/− | − | − | Stable | Stable |
| 31 | −/− | −/− | −/− | − | − | Stable | Stable |
| 32 | +/+ | +/+ | −/+ FN | − | − | High | High |
| 33 | −/− | −/− | −/− | − | − | Stable | Stable |
| 34 | +/+ | +/+ | −/+ FN | + | − | High | High |
| 35 | −/− | −/− | −/− | − | − | Stable | Stable |
| 36 | −/− | −/− | −/− | − | − | Stable | Stable |
| 37 | +/+ | +/+ | +/− FP | + | + | High | High |
| 38 | −/− | −/− | −/− | − | − | Stable | Stable |
| 39 | −/− | −/− | −/− | − | − | Stable | Stable |
| 40 | −/− | −/− | −/− | − | − | Stable | Stable |
| 41 | −/− | −/− | −/− | − | − | Stable | Stable |
| 42 | +/+ | +/+ | +/+ | + | − | High | High |
| 43 | +/+ | +/+ | +/− FP | + | + | High | High |
| 44 | −/− | −/− | −/− | − | − | Stable | Stable |
| Cases | BAT25 (M) (NGS/Multiplex PCR) | BAT26 (M) (NGS/Multiplex PCR ) | MSI status (NGS) | MSI status (Multiplex PCR) |
|---|---|---|---|---|
| 1 | +/+ | +/+ | High | High |
| 2 | +/+ | +/+ | High | High |
| 3 | −/− | −/− | Stable | Stable |
| 4 | +/+ | +/+ | High | High |
| 5 | −/− | −/− | Stable | Low |
| 6 | −/− | −/− | Stable | Stable |
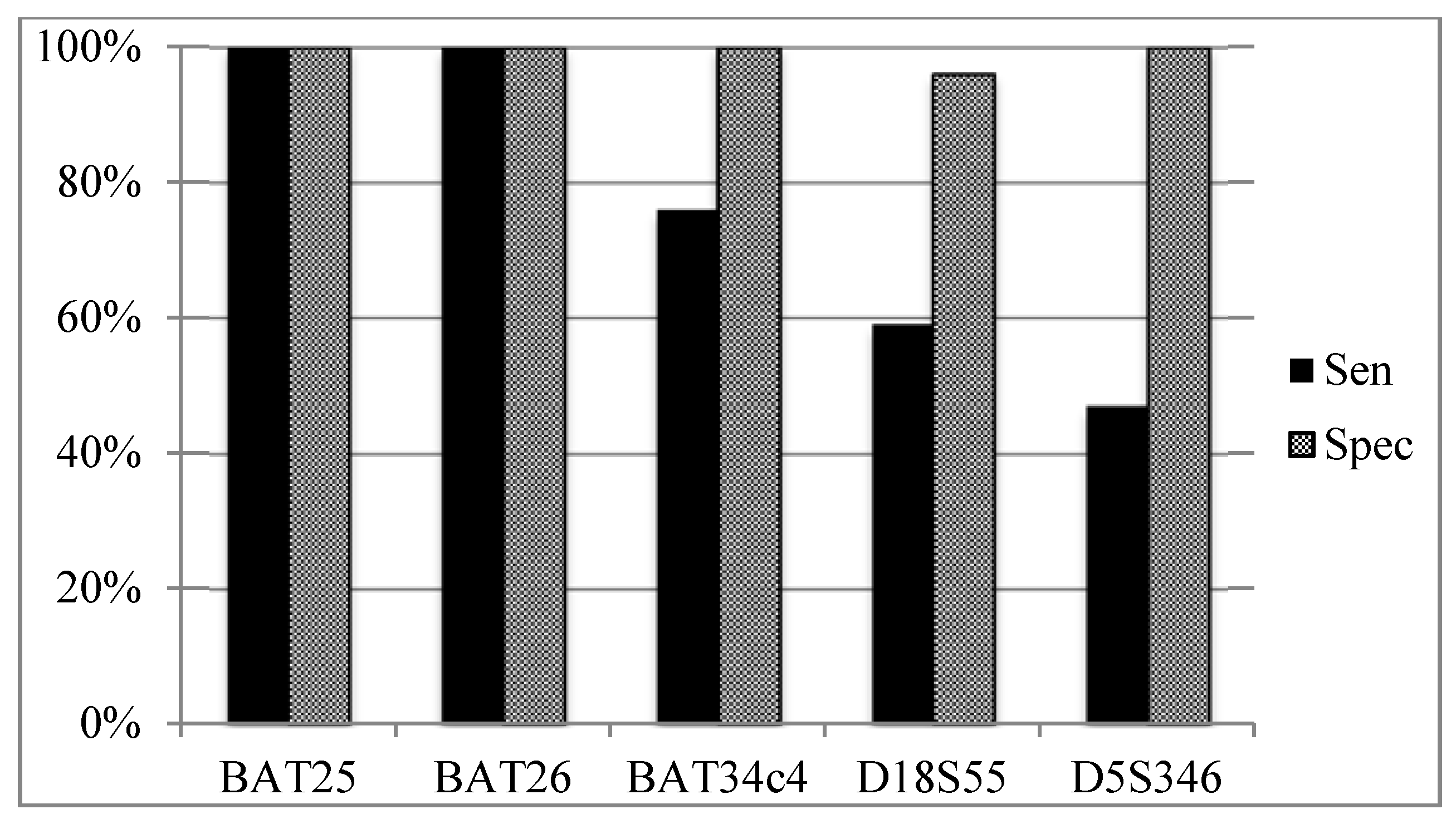
3.5. Sensitivity and Specificity of Individual MSI Loci According to Overall MSI Status (Figure 4)



4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgement
Author Contributions
Conflict of interest
References
- Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: Genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet. Med. 2009, 11, 35–41. [Google Scholar]
- Woerner, S.M.; Kloor, M.; Mueller, A.; Rueschoff, J.; Friedrichs, N.; Buettner, R.; Buzello, M.; Kienle, P.; Knaebel, H.-P.; Kunstmann, E.; et al. Microsatellite instability of selective target genes in HNPCC-associated colon adenomas. Oncogene 2005, 24, 2523–2535. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Cicek, M.S.; Lindor, N.M.; Gallinger, S.; Bapat, B.; Hopper, J.L.; Jenkins, M.A.; Young, J.; Buchanan, D.; Walsh, M.D.; le Marchand, L.; et al. Quality assessment and correlation of microsatellite instability and immunohistochemical markers among population- and clinic-based colorectal tumors results from the Colon Cancer Family Registry. J. Mol. Diagn. 2011, 13, 27–281. [Google Scholar] [CrossRef]
- Deschoolmeester, V.; Baay, M.; Wuyts, W.; van Marck, E.; van Damme, N.; Vermeulen, P.; Lukaszuk, K.; Lardon, F.; Vermorken, J.B. Detection of microsatellite instability in colorectal cancer using an alternative multiplex assay of quasi-monomorphic mononucleotide markers. J. Mol. Diagn. 2008, 10, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Pastrello, C.; Baglioni, S.; Tibiletti, M.G.; Papi, L.; Fornasarig, M.; Morabito, A.; Agostini, M.; Genuardi, M.; Viel, A. Stability of BAT26 in tumours of hereditary nonpolyposis colorectal cancer patients with MSH2 intragenic deletion. Eur. J. Hum. Genet. 2006, 14, 63–68. [Google Scholar] [PubMed]
- Schofield, L.; Watson, N.; Grieu, F.; Li, W.Q.; Zeps, N.; Harvey, J.; Stewart, C.; Abdo, M.; Goldblatt, J.; Iacopetta, B. Population-based detection of Lynch syndrome in young colorectal cancer patients using microsatellite instability as the initial test. Int. J. Cancer 2009, 124, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2003, 349, 247. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M.; Smith, E.J.; Behling, C.A.; Nguyen, L.; Tajima, A.; Doctolero, R.T.; Cabrera, B.L.; Goel, A.; Arnold, C.A.; Miyai, K.; et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology 2004, 126, 394. [Google Scholar] [CrossRef] [PubMed]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 3219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J. Era of universal testing of microsatellite instability in colorectal cancer. World J. Gastrointest Oncol. 2013, 5, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.W.; Cannon, J.A.; David, D.S.; Early, D.S.; Ford, J.M.; Giardiello, F.M.; Halverson, A.L.; Hamilton, S.R.; Hampel, H.; Ismail, M.K.; et al. Colorectal cancer screening. J. Natl. Compr. Canc. Netw. 2013, 11, 1538–1575. [Google Scholar] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Ni, H.; He, Y.; Pong, R.; Lin, P.; Lu, L.H.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012. Article ID 251364. [Google Scholar]
- Mciver, L.J.; Fonville, N.C.; Karunasena, E.; Garner, H.R. Microsatellite genotyping reveals a signature in breast cancer exomes. Breast Cancer Res. Treat 2014, 145, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; Scroggins, S.M.; Hampel, H.L.; Turner, E.H.; Pritchard, C.C. Microsatellite instability detection by next generation sequening. Clin. Chem. 2014, 60, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Laird, P.W.; Park, P.J. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 2013, 155, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.H.; Soong, T.D.; Elemento, O. A novel approach for characterizing microsatellite instability in cancer cells. PLOS ONE 2013, 8, e63056. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.; Lee, S.; Han, T.; Moon, S.Y.; Yun, M.S.; Kong, S.; Jho, S.; Choe, J.; Yu, J.; Lee, H.; et al. Comprehensive genome- and transcriptome-wide analyses of mutations associated with microsatellite instability in Korean gastric cancers. Genome Res. 2013, 23, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.K.; Nickerson, E.; Simons, J.F.; Janne, P.A.; Tengs, T.; Yuza, Y.; Garraway, L.A.; LaFramboise, T.; Lee, J.C.; Shah, K.; et al. Sensitive mutation de-tection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat. Med. 2006, 12, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.; Beshay, V.; Peter MacCallum Cancer Institute: East Melbourne, Victoria, Australia. Unpublished data. 2010.
- Waring, P.; Cliff, S.; University of Melbourne, Parkville, Victoria, Australia. Unpublished data. 2013.
- Luthra, R.; Patel, K.P.; Reddy, N.G.; Haghshenas, V.; Routbort, M.J.; Harmon, M.A.; Barkoh, B.A.; Kanagal-Shamanna, R.; Ravandi, F.; Cortes, J.E.; et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: Applicability for diagnostics and disease monitoring. Haematologica 2014, 99, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Buckton, A.J.; Wilkinson, S.L.; John, S.; Walsh, R.; Novotny, T.; Valaskova, I.; Gupta, M.; Game, L.; Barton, P.J.; et al. Towards clinical molecular diagnosis of inherited cardiac conditions: A comparison of bench-top genome DNA sequencers. PLOS ONE 2013, 8, e67744. [Google Scholar] [CrossRef] [PubMed]
- Kanagal-Shamanna, R.; Portier, B.P.; Singh, R.R.; Routbort, M.J.; Aldape, K.D.; Handal, B.A.; Rahimi, H.; Reddy, N.G.; Barkoh, B.A.; Mishra, B.M.; et al. Next-generation sequencing-based multi-gene mutation profiling of solid tumors using fine needle aspiration samples: Promises and challenges for routine clinical diagnostics. Mod. Pathol. 2014, 27, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.P.; Hoang, J.M.; Li, Y.J.; Seruca, R.; Carneiro, F.; Sobrinho-Simoes, M.; Lothe, R.A.; Gleeson, C.M.; Russell, S.E.; Muzeau, F.; et al. Determination of the replication error phenotype in human tumors without the requirement for matching normal DNA by analysis of mononucleotide repeat microsatellites. Genes Chromosomes Cancer 1998, 21, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Brennetot, C.; Buhard, O.; Jourdan, F.; Flejou, J.; Duval, A.; Hamelin, R. Mononucleotide repeats BAT-26 and BAT-25 accurately detect MSI-H tumors and predict tumor content: implications for population screening. Int. J. Cancer 2005, 113, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Mead, L.J.; Jenkins, M.A.; Young, J.; Royce, S.G.; Smith, L.; St John, D.J.; Macrae, F.; Giles, G.G.; Hopper, J.L.; Southey, M.C. Microsatellite instability markers for identifying early-onset colorectal cancers caused by germ-line mutations in DNA mismatch repair genes. Clin. Cancer Res. 2007, 13, 2865–2869. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gan, C.; Love, C.; Beshay, V.; Macrae, F.; Fox, S.; Waring, P.; Taylor, G. Applicability of Next Generation Sequencing Technology in Microsatellite Instability Testing. Genes 2015, 6, 46-59. https://doi.org/10.3390/genes6010046
Gan C, Love C, Beshay V, Macrae F, Fox S, Waring P, Taylor G. Applicability of Next Generation Sequencing Technology in Microsatellite Instability Testing. Genes. 2015; 6(1):46-59. https://doi.org/10.3390/genes6010046
Chicago/Turabian StyleGan, Chun, Clare Love, Victoria Beshay, Finlay Macrae, Stephen Fox, Paul Waring, and Graham Taylor. 2015. "Applicability of Next Generation Sequencing Technology in Microsatellite Instability Testing" Genes 6, no. 1: 46-59. https://doi.org/10.3390/genes6010046
APA StyleGan, C., Love, C., Beshay, V., Macrae, F., Fox, S., Waring, P., & Taylor, G. (2015). Applicability of Next Generation Sequencing Technology in Microsatellite Instability Testing. Genes, 6(1), 46-59. https://doi.org/10.3390/genes6010046