
In Silico Characterization of miRNA and Long Non-Coding RNA Interplay in Multiple Myeloma

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Expression Profiling
2.3. Statistical Analysis
3. Results
3.1. lncRNA and miRNA Interplay in MM
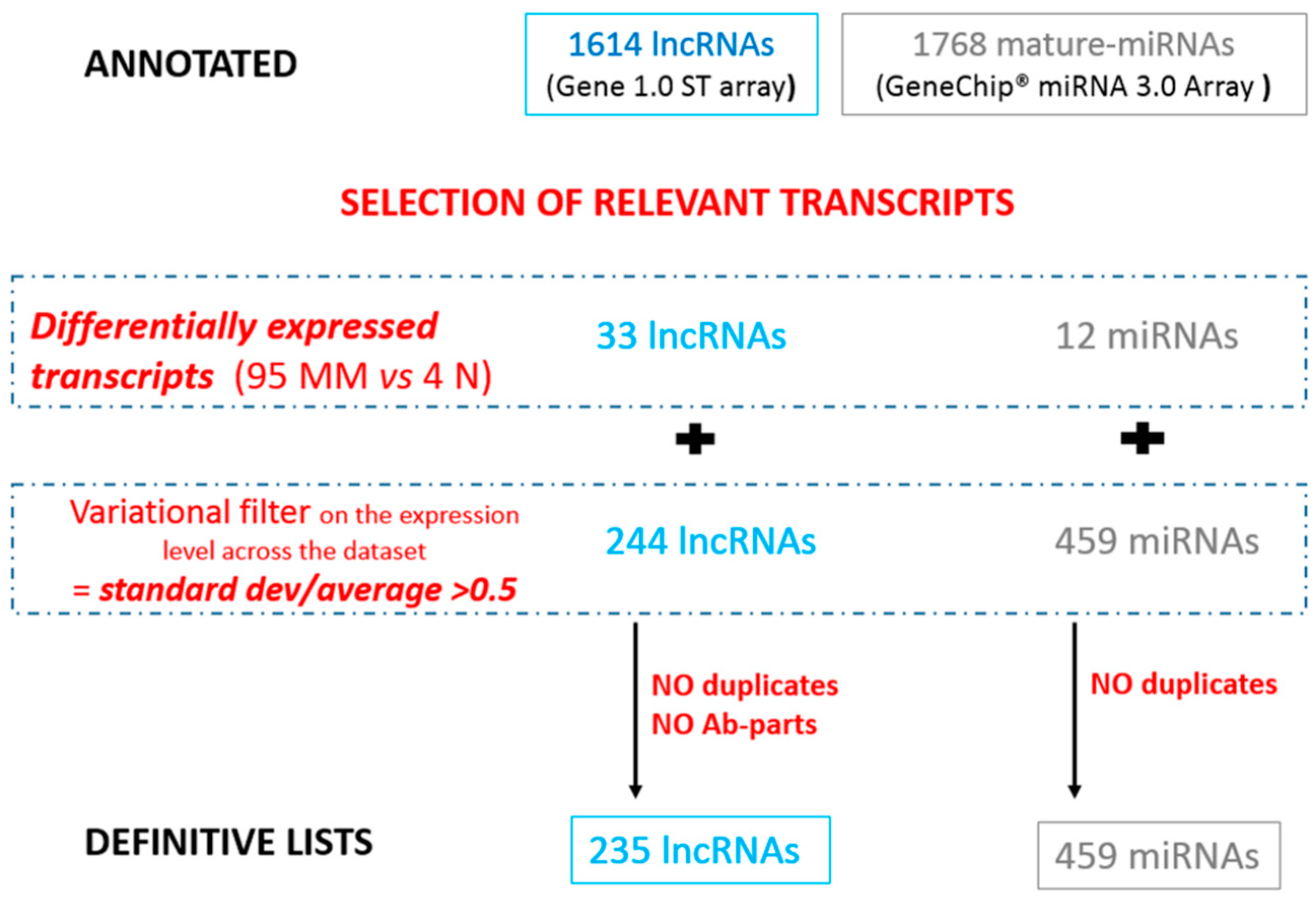
3.1.1. Selection of Relevant lncRNAs and miRNAs
3.1.2. Identification of lncRNAs miRNA-Target
3.1.3. Correlation between lncRNAs and Corresponding miRNA Targets in MM
3.2. lncRNA and miRNA Interplay in PCL
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, L.; Tassone, P.; Neri, A. Molecular profiling of multiple myeloma: From gene expression analysis to next-generation sequencing. Expert. Opin. Biol. Ther. 2013, 13, S55–S68. [Google Scholar] [CrossRef] [PubMed]
- Calura, E.; Bisognin, A.; Manzoni, M.; Todoerti, K.; Taiana, E.; Sales, G.; Morgan, G.J.; Tonon, G.; Amodio, N.; Tassone, P.; et al. Disentangling the microRNA regulatory milieu in multiple myeloma: Integrative genomics analysis outlines mixed miRNA-TF circuits and pathway-derived networks modulated in t(4;14) patients. Oncotarget 2016, 7, 2367–2378. [Google Scholar] [PubMed]
- Gutierrez, N.C.; Sarasquete, M.E.; Misiewicz-Krzeminska, I.; Delgado, M.; De Las, R.J.; Ticona, F.V.; Ferminan, E.; Martin-Jimenez, P.; Chillon, C.; Risueno, A.; et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia 2010, 24, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Biasiolo, M.; Agnelli, L.; Todoerti, K.; Mosca, L.; Fabris, S.; Sales, G.; Deliliers, G.L.; Bicciato, S.; Lombardi, L.; et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood 2009, 114, e20–e26. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Agnelli, L.; Lombardi, L.; Tassone, P.; Neri, A. MicroRNAs in the pathobiology of multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, L.; Barlogie, B.; Stephens, O.; Wu, X.; Williams, D.R.; Cartron, M.A.; van, R.F.; Nair, B.; Waheed, S.; et al. High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. Proc. Natl. Acad. Sci. USA 2010, 107, 7904–7909. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A brief review on the mechanisms of miRNA regulation. Genom. Proteom. Bioinf. 2009, 7, 147–154. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Karreth, F.A.; Pandolfi, P.P. ceRNA cross-talk in cancer: When ce-bling rivalries go awry. Cancer Discov. 2013, 3, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; Garcia, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Z.; Jiang, J.; Xu, C.; Kang, J.; Xiao, L.; Wu, M.; Xiong, J.; Guo, X.; Liu, H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev. Cell 2013, 25, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef] [PubMed]
- LNCipedia repository. Available online: http://www.lncipedia.org/ (accessed on 1 August 2016).
- van, H.S.; van, I.M.; Jacobi, J.; Boymans, S.; Essers, P.B.; de, B.E.; Hao, W.; MacInnes, A.W.; Cuppen, E.; Simonis, M. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genom. Biol. 2014, 15, R6–R17. [Google Scholar]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Reczko, M.; Maragkakis, M.; Dalamagas, T.M.; Hatzigeorgiou, A.G. DIANA-LncBase: Experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013, 41, D239–D245. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejias, A.; Tay, Y. Competing endogenous RNA networks: Tying the essential knots for cancer biology and therapeutics. J. Hematol. Oncol. 2015, 8, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Paci, P.; Colombo, T.; Farina, L. Computational analysis identifies a sponge interaction network between long non-coding RNAs and messenger RNAs in human breast cancer. BMC Syst. Biol. 2014, 8, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, J.; Wang, W. Construction and investigation of breast-cancer-specific ceRNA network based on the mRNA and miRNA expression data. IET Syst. Biol. 2014, 8, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Liao, Q.; Jiang, X.; Shao, Y.; Xiao, B.; Xi, Y.; Guo, J. Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci. Rep. 2014, 4, 6088–6094. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.C.; Hsiao, T.H.; Chen, Y.; Chuang, E.Y. Parameter optimization for constructing competing endogenous RNA regulatory network in glioblastoma multiforme and other cancers. BMC Genomics 2015, 16, S1–S13. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, D.; Agnelli, L.; Taiana, E.; Galletti, S.; Manzoni, M.; Todoerti, K.; Musto, P.; Strozzi, F.; Neri, A. Distinct lncRNA transcriptional fingerprints characterize progressive stages of multiple myeloma. Oncotarget 2016, 7, 14814–14830. [Google Scholar] [PubMed]
- Malek, E.; Kim, B.G.; Driscoll, J.J. Identification of Long Non-Coding RNAs Deregulated in Multiple Myeloma Cells Resistant to Proteasome Inhibitors. Genes 2016, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, L.; Fabris, S.; Bicciato, S.; Basso, D.; Baldini, L.; Morabito, F.; Verdelli, D.; Todoerti, K.; Lambertenghi-Deliliers, G.; Lombardi, L.; et al. Upregulation of translational machinery and distinct genetic subgroups characterise hyperdiploidy in multiple myeloma. Br. J. Haematol. 2007, 136, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Todoerti, K.; Agnelli, L.; Fabris, S.; Lionetti, M.; Tuana, G.; Mosca, L.; Lombardi, L.; Grieco, V.; Bianchino, G.; D′Auria, F.; et al. Transcriptional characterization of a prospective series of primary plasma cell leukemia revealed signatures associated with tumor progression and poorer outcome. Clin. Cancer Res. 2013, 19, 3247–3258. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- MikeJSeo/SAM. Available online: https://github.com/MikeJSeo/SAM (accessed on 1 August 2016).
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, L.; Bicciato, S.; Mattioli, M.; Fabris, S.; Intini, D.; Verdelli, D.; Baldini, L.; Morabito, F.; Callea, V.; Lombardi, L.; et al. Molecular classification of multiple myeloma: A distinct transcriptional profile characterizes patients expressing CCND1 and negative for 14q32 translocations. J. Clin. Oncol. 2005, 23, 7296–7306. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, D.; Todoerti, K.; Tuana, G.; Agnelli, L.; Mosca, L.; Lionetti, M.; Fabris, S.; Colapietro, P.; Miozzo, M.; Ferrarini, M.; et al. The expression pattern of small nucleolar and small Cajal body-specific RNAs characterizes distinct molecular subtypes of multiple myeloma. Blood Cancer J. 2012, 2, e96–e103. [Google Scholar] [CrossRef] [PubMed]
- Deng, K.; Wang, H.; Guo, X.; Xia, J. The cross talk between long, non-coding RNAs and microRNAs in gastric cancer. Acta Biochim. Biophys. Sin. 2016, 48, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Haecker, I.; Gay, L.A.; Yang, Y.; Hu, J.; Morse, A.M.; McIntyre, L.M.; Renne, R. Ago HITS-CLIP expands understanding of Kaposi′s sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog. 2012, 8, e1002884–e1002900. [Google Scholar] [CrossRef] [PubMed]
- Karginov, F.V.; Hannon, G.J. Remodeling of Ago2-mRNA interactions upon cellular stress reflects miRNA complementarity and correlates with altered translation rates. Genes Dev. 2013, 27, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.N.; Khong, T.; Segal, D.; Yao, Y.; Riffkin, C.D.; Garnier, J.M.; Khaw, S.L.; Lessene, G.; Spencer, A.; Herold, M.J.; et al. Hierarchy for targeting pro-survival BCL2 family proteins in multiple myeloma: Pivotal role of MCL1. Blood 2016, 128, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Simpson, H.M.; Khan, R.Z.; Song, C.; Sharma, D.; Sadashivaiah, K.; Furusawa, A.; Liu, X.; Nagaraj, S.; Sengamalay, N.; Sadzewicz, L.; et al. Concurrent Mutations in ATM and Genes Associated with Common gamma Chain Signaling in Peripheral T Cell Lymphoma. PLoS ONE 2015, 10, e0141906–e0141922. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Sheng, Y.; Ding, S.; Chen, J.; Tan, X.; Zeng, T.; Qin, D.; Zhu, L.; Huang, A.; Tang, H. LINC00052 regulates the expression of NTRK3 by miR-128 and miR-485–3p to strengthen HCC cells invasion and migration. Oncotarget 2016, 7, 47593–47608. [Google Scholar] [CrossRef] [PubMed]
- Guidi, M.; Muinos-Gimeno, M.; Kagerbauer, B.; Marti, E.; Estivill, X.; Espinosa-Parrilla, Y. Overexpression of miR-128 specifically inhibits the truncated isoform of NTRK3 and upregulates BCL2 in SH-SY5Y neuroblastoma cells. BMC Mol. Biol. 2010, 11, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Rossi, M.; Caracciolo, D.; Gulla, A.; Tagliaferri, P.; Tassone, P. Mir-221/222 are promising targets for innovative anticancer therapy. Expert. Opin. Ther. Targets 2016, 20, 1099–1108. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| lncRNA | lncRNA Chr. 1 | miRNA | miRNA Chr. 1 | Correlation | Target Prediction 2 | ||
|---|---|---|---|---|---|---|---|
| Corr. Coeff. | q-val. | lncRNA Target Sequence from/to | q-val. | ||||
| DLEU2:26 | 13q14 | mir-3175 | 15q26 | −0.35 | 0.0444 | 668–690 | 0.011 |
| KLF3-AS1:2 | 4p14 | mir-4787-5p | 3p21 | −0.36 | 0.0319 | 1890–1911 | 0.045 |
| LINC00173:6 | 12q24 | mir-221-3p | Xp11 | −0.49 | 0.0003 | 2485–2506 | 0.013 |
| LINC00173:9 | 12q24 | mir-221-3p | Xp11 | −0.49 | 0.0003 | 10737–10759 | 0.019 |
| AGBL1-4 | 15q25.3 | mir-185-5p | 22q11 | −0.37 | 0.0266 | 3303–3321 | 0.040 |
| MCL1-2:1 | 1q21 | mir-106a-5p | Xq26 | −0.36 | 0.0351 | 5285–5306 | <1E-06 |
| MCL1-2:1 | 1q21 | mir-18a-5p | 13q31 | −0.43 | 0.0041 | 4377–4397 | 0.043 |
| MCL1-2:1 | 1q21 | mir-18b-5p | Xq26 | −0.35 | 0.0388 | 2903–2923 | 0.023 |
| MCL1-2:1 | 1q21 | mir-20a-5p | 13q31 | −0.36 | 0.0351 | 98–120 | 0.047 |
| MCL1-2:1 | 1q21 | mir-17-5p | 13q31 | −0.35 | 0.0449 | 184–206 | 0.011 |
| WDR73-3:10 | 15q25.2 | mir-423-5p | 17q11 | −0.40 | 0.0124 | 2090–2113 | 0.049 |
| miRNA | lncRNA Transcript | Corr. Coeff. | p-val. |
|---|---|---|---|
| hsa-mir-185-5p | lnc-AGBL1-4:1 | −0.451 | 0.012 |
| hsa-mir-106a-5p | lnc-MCL1-2:1 | −0.377 | 0.039 |
| hsa-mir-18a-5p | lnc-MCL1-2:1 | −0.431 | 0.017 |
| hsa-mir-18b-5p | lnc-MCL1-2:1 | −0.466 | 0.009 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronchetti, D.; Manzoni, M.; Todoerti, K.; Neri, A.; Agnelli, L. In Silico Characterization of miRNA and Long Non-Coding RNA Interplay in Multiple Myeloma. Genes 2016, 7, 107. https://doi.org/10.3390/genes7120107
Ronchetti D, Manzoni M, Todoerti K, Neri A, Agnelli L. In Silico Characterization of miRNA and Long Non-Coding RNA Interplay in Multiple Myeloma. Genes. 2016; 7(12):107. https://doi.org/10.3390/genes7120107
Chicago/Turabian StyleRonchetti, Domenica, Martina Manzoni, Katia Todoerti, Antonino Neri, and Luca Agnelli. 2016. "In Silico Characterization of miRNA and Long Non-Coding RNA Interplay in Multiple Myeloma" Genes 7, no. 12: 107. https://doi.org/10.3390/genes7120107
APA StyleRonchetti, D., Manzoni, M., Todoerti, K., Neri, A., & Agnelli, L. (2016). In Silico Characterization of miRNA and Long Non-Coding RNA Interplay in Multiple Myeloma. Genes, 7(12), 107. https://doi.org/10.3390/genes7120107