
The Future is The Past: Methylation QTLs in Schizophrenia

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Genetic Architecture of SCZ
3. The GWAS Era
4. In Search of Function
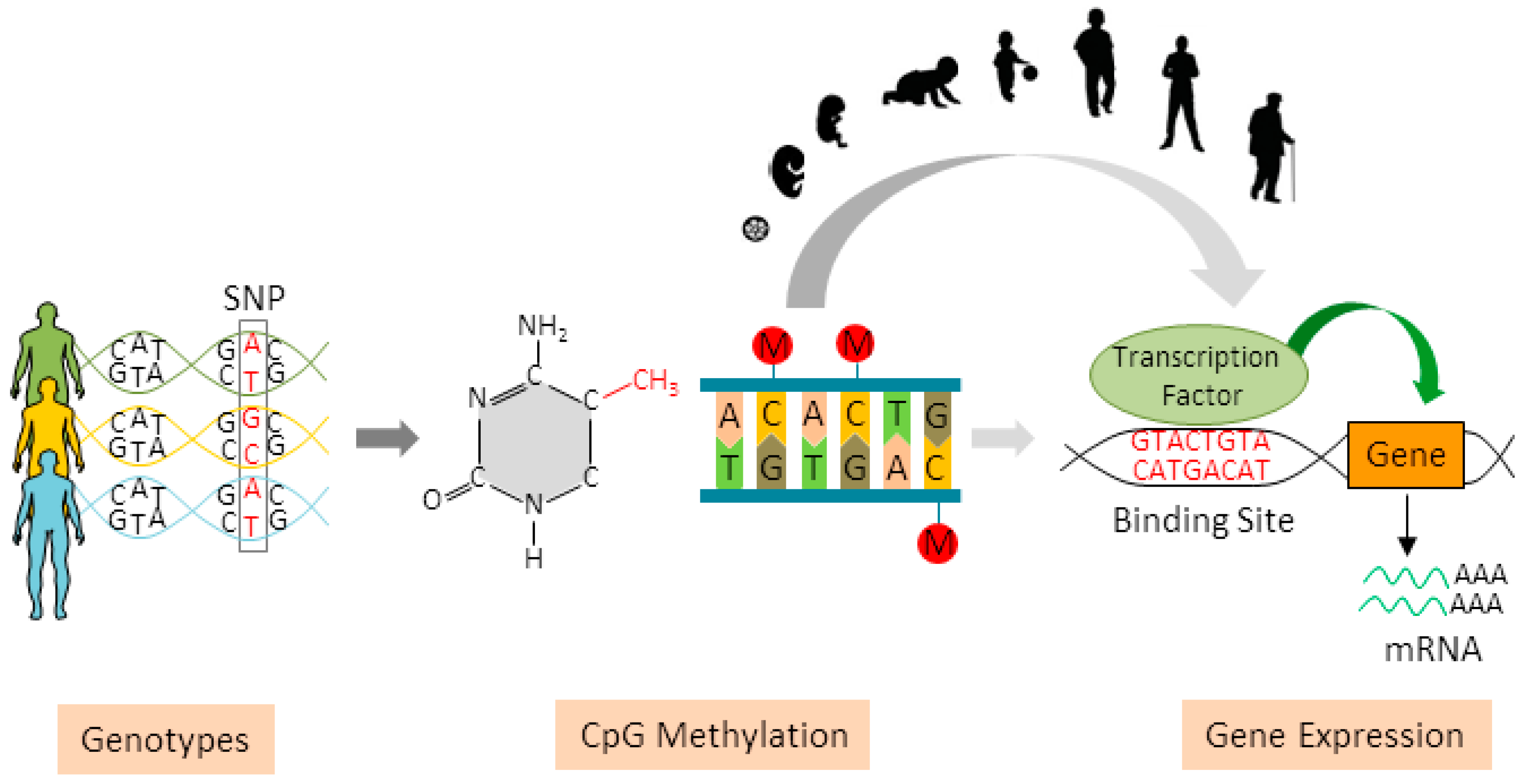
5. Dynamic DNA Methylation
6. What Are meQTLs?
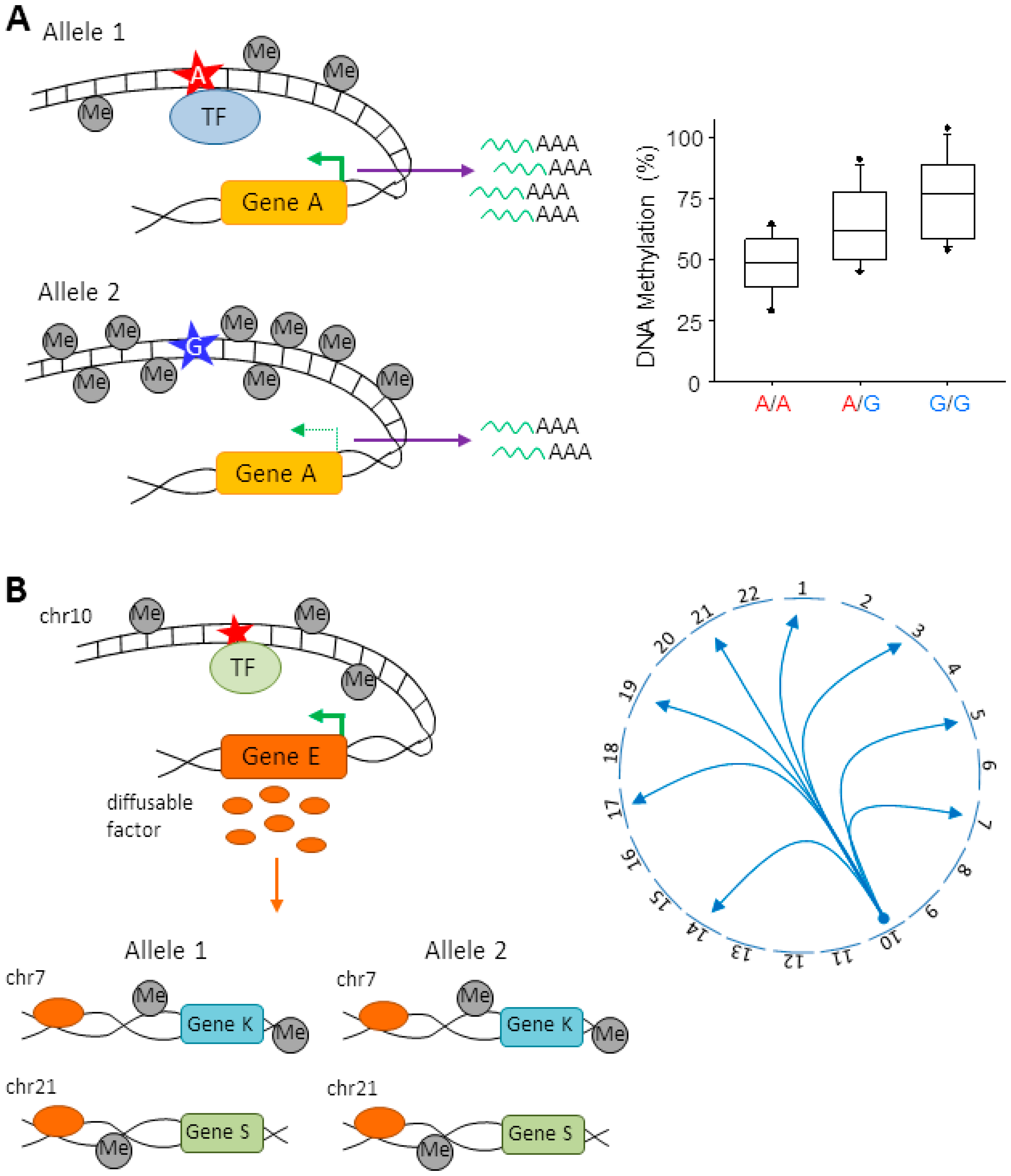
6.1. Local meQTLs
6.2. Distant meQTLs
7. Molecular Mechanisms for meQTLs
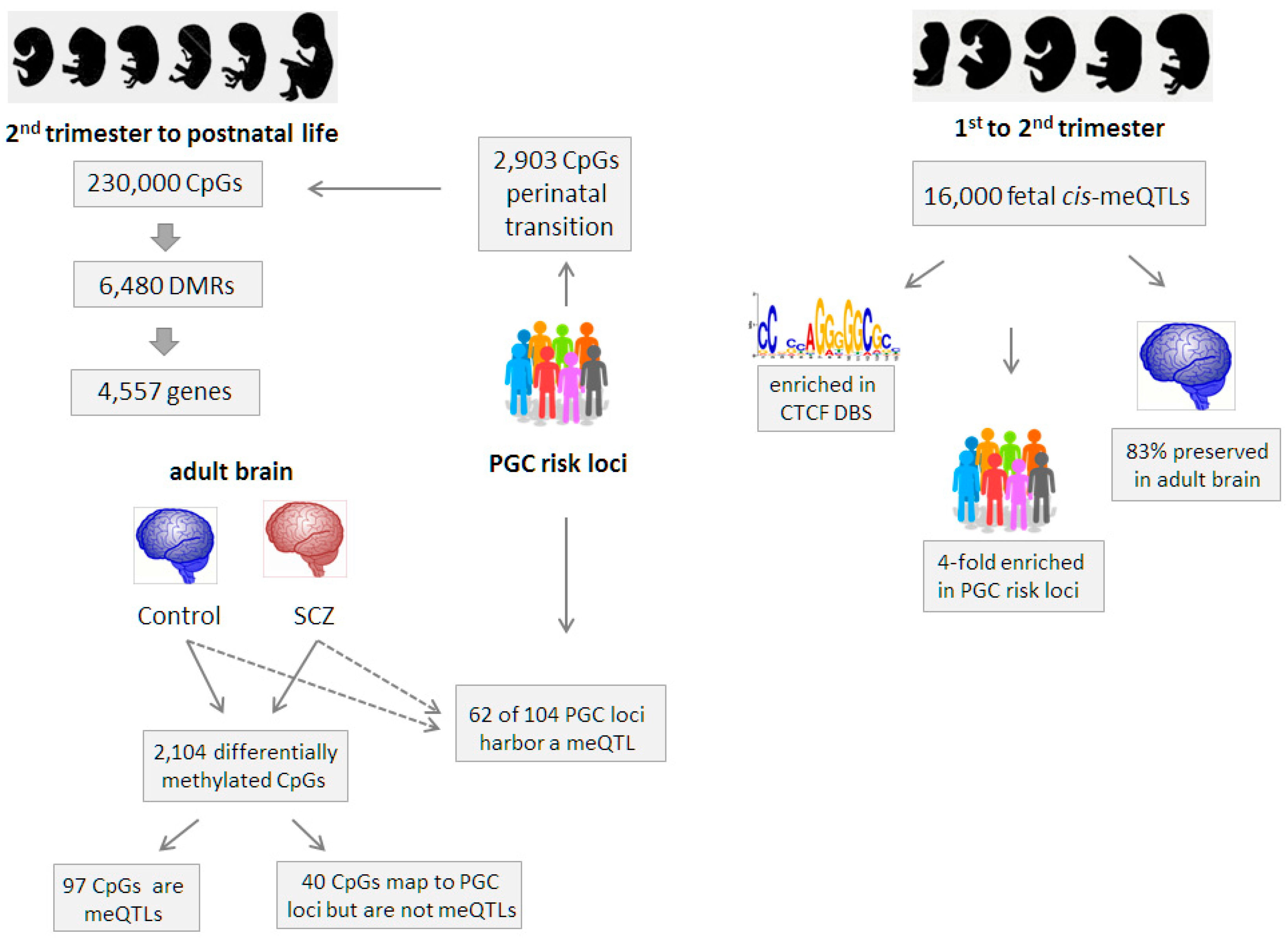
8. Early meQTL Studies in the Brain
9. meQTLs Are Enriched at Regulatory Sites
10. meQTLs in SCZ
11. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gejman, P.V.; Sanders, A.R.; Duan, J. The role of genetics in the etiology of schizophrenia. Psychiatr. Clin. N. Am. 2010, 33, 35–66. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.F.; Daly, M.J.; O’Donovan, M. Genetic architectures of psychiatric disorders: The emerging picture and its implications. Nat. Rev. Genet. 2012, 13, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, P.; Yip, B.H.; Björk, C.; Pawitan, Y.; Cannon, T.D.; Sullivan, P.F.; Hultman, C.M. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: A population-based study. Lancet 2009, 373, 234–239. [Google Scholar] [CrossRef]
- Wray, N.R.; Gottesman, I.I. Using summary data from the danish national registers to estimate heritabilities for schizophrenia, bipolar disorder, and major depressive disorder. Front. Genet. 2012, 3, 118. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.F.; Kendler, K.S.; Neale, M.C. Schizophrenia as a complex trait: Evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 2003, 60, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Jablensky, A. Epidemiology of schizophrenia: The global burden of disease and disability. Eur. Arch. Psychiatr. Clin. Neurosci. 2000, 250, 274–285. [Google Scholar] [CrossRef]
- Kim, Y.; Zerwas, S.; Trace, S.E.; Sullivan, P.F. Schizophrenia genetics: Where next? Schizophr. Bull. 2011, 37, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; O’Dushlaine, C.; Chambert, K.; Moran, J.L.; Kähler, A.K.; Akterin, S.; Bergen, S.E.; Collins, A.L.; Crowley, J.J.; Fromer, M.; et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013, 45, 1150–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreassen, O.A.; Thompson, W.K.; Schork, A.J.; Ripke, S.; Mattingsdal, M.; Kelsoe, J.R.; Kendler, K.S.; O’Donovan, M.C.; Rujescu, D.; Werge, T.; et al. Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS Genet. 2013, 9, e1003455. [Google Scholar] [CrossRef] [PubMed]
- BrainSeq: A Human Brain Genomics Consortium. BrainSeq: Neurogenomics to drive novel target discovery for neuropsychiatric disorders. Neuron 2015, 88, 1078–1083. [Google Scholar]
- Gandal, M.J.; Leppa, V.; Won, H.; Parikshak, N.N.; Geschwind, D.H. The road to precision psychiatry: Translating genetics into disease mechanisms. Nat. Neurosci. 2016, 19, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [Green Version]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [Green Version]
- Kichaev, G.; Pasaniuc, B. Leveraging functional-annotation data in trans-ethnic Fine-mapping studies. Am. J. Hum. Genet. 2015, 97, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Willer, C.; Sanna, S.; Abecasis, G. Genotype imputation. Annu. Rev. Genom. Hum. Genet. 2009, 10, 387–406. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [Green Version]
- Freedman, M.L.; Monteiro, A.N.A.; Gayther, S.A.; Coetzee, G.A.; Risch, A.; Plass, C.; Casey, G.; De Biasi, M.; Carlson, C.; Duggan, D.; et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet. 2011, 43, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Howie, B.N.; Donnelly, P.; Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009, 5, e1000529. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Willer, C.J.; Ding, J.; Scheet, P.; Abecasis, G.R. MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010, 34, 816–834. [Google Scholar] [CrossRef] [PubMed]
- Dimas, A.S.; Deutsch, S.; Stranger, B.E.; Montgomery, S.B.; Borel, C.; Attar-Cohen, H.; Ingle, C.; Beazley, C.; Gutierrez Arcelus, M.; Sekowska, M.; et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 2009, 325, 1246–1250. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Seo, J.-H.; Stranger, B.; McKenna, A.; Pe’er, I.; Laframboise, T.; Brown, M.; Tyekucheva, S.; Freedman, M.L. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 2013, 152, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Kasowski, M.; Grubert, F.; Heffelfinger, C.; Hariharan, M.; Asabere, A.; Waszak, S.M.; Habegger, L.; Rozowsky, J.; Shi, M.; Urban, A.E.; et al. Variation in transcription factor binding among humans. Science 2010, 328, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Degner, J.F.; Pai, A.A.; Pique-Regi, R.; Veyrieras, J.-B.; Gaffney, D.J.; Pickrell, J.K.; De Leon, S.; Michelini, K.; Lewellen, N.; Crawford, G.E.; et al. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science 2013, 342, 744–747. [Google Scholar]
- McVicker, G.; van de Geijn, B.; Degner, J.F.; Cain, C.E.; Banovich, N.E.; Raj, A.; Lewellen, N.; Myrthil, M.; Gilad, Y.; Pritchard, J.K. Identification of genetic variants that affect histone modifications in human cells. Science 2013, 342, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Fridley, B.L.; Song, H.; Lawrenson, K.; Cunningham, J.M.; Ramus, S.J.; Cicek, M.S.; Tyrer, J.; Stram, D.; Larson, M.C.; et al. Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat. Commun. 2013, 4, 1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.-Y.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.I.; van de Geijn, B.; Raj, A.; Knowles, D.A.; Petti, A.A.; Golan, D.; Gilad, Y.; Pritchard, J.K. RNA splicing is a primary link between genetic variation and disease. Science 2016, 352, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L. Epigenetics; Garland Science: New York, NY, USA, 2014. [Google Scholar]
- Schübeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.D.; Smith, A.J.H.; De Gobbi, M.; Flenley, M.; Hughes, J.R.; Vernimmen, D.; Ayyub, H.; Sharpe, J.A.; Sloane-Stanley, J.A.; Sutherland, L.; et al. An interspecies analysis reveals a key role for unmethylated CpG dinucleotides in vertebrate Polycomb complex recruitment. EMBO J. 2012, 31, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Tanay, A.; O’Donnell, A.H.; Damelin, M.; Bestor, T.H. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc. Natl. Acad. Sci. USA 2007, 104, 5521–5526. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Spengler, D. DNA memories of early social life. Neuroscience 2014, 264, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.C.; Nap, J.P. Genetical genomics: The added value from segregation. Trends Genet. 2001, 17, 388–391. [Google Scholar] [CrossRef]
- Rockman, M.V.; Kruglyak, L. Genetics of global gene expression. Nat. Rev. Genet. 2006, 7, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Tycko, B. Mapping allele-specific DNA methylation: A new tool for maximizing information from GWAS. Am. J. Hum. Genet. 2010, 86, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Klengel, T.; Mehta, D.; Anacker, C.; Rex-Haffner, M.; Pruessner, J.C.; Pariante, C.M.; Pace, T.W.W.; Mercer, K.B.; Mayberg, H.S.; Bradley, B.; et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 2013, 16, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Turecki, G.; Meaney, M.J. Effects of the social environment and stress on glucocorticoid receptor gene methylation: A systematic review. Biol. Psychiatry 2016, 79, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Turecki, G.; Ota, V.K.; Belangero, S.I.; Jackowski, A.; Kaufman, J. Early life adversity, genomic plasticity, and psychopathology. Lancet Psychiatry 2014, 1, 461–466. [Google Scholar] [CrossRef]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12, R10. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.T.; Tsai, P.-C.; Yang, T.-P.; Pidsley, R.; Nisbet, J.; Glass, D.; Mangino, M.; Zhai, G.; Zhang, F.; Valdes, A.; et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012, 8, e1002629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drong, A.W.; Nicholson, G.; Hedman, A.K.; Meduri, E.; Grundberg, E.; Small, K.S.; Shin, S.-Y.; Bell, J.T.; Karpe, F.; Soranzo, N.; et al. The presence of methylation quantitative trait loci indicates a direct genetic influence on the level of DNA methylation in adipose tissue. PLoS ONE 2013, 8, e55923. [Google Scholar] [CrossRef] [PubMed]
- Gertz, J.; Varley, K.E.; Reddy, T.E.; Bowling, K.M.; Pauli, F.; Parker, S.L.; Kucera, K.S.; Willard, H.F.; Myers, R.M. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011, 7, e1002228. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.R.; van der Brug, M.P.; Hernandez, D.G.; Traynor, B.J.; Nalls, M.A.; Lai, S.-L.; Arepalli, S.; Dillman, A.; Rafferty, I.P.; Troncoso, J.; et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010, 6, e1000952. [Google Scholar] [CrossRef] [PubMed]
- Hannon, E.; Spiers, H.; Viana, J.; Pidsley, R.; Burrage, J.; Murphy, T.M.; Troakes, C.; Turecki, G.; O’Donovan, M.C.; Schalkwyk, L.C.; et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat. Neurosci. 2016, 19, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.E.; Gao, Y.; Deep-Soboslay, A.; Tao, R.; Hyde, T.M.; Weinberger, D.R.; Kleinman, J.E. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci. 2016, 19, 40–47. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar]
- Del Rosario, R.C.-H.; Poschmann, J.; Rouam, S.L.; Png, E.; Khor, C.C.; Hibberd, M.L.; Prabhakar, S. Sensitive detection of chromatin-altering polymorphisms reveals autoimmune disease mechanisms. Nat. Methods 2015, 12, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Westra, H.-J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, A.D.; Hu, M.; Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nat. Rev. Mol. Cell Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lienert, F.; Wirbelauer, C.; Som, I.; Dean, A.; Mohn, F.; Schübeler, D. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 2011, 43, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Banovich, N.E.; Lan, X.; McVicker, G.; van de Geijn, B.; Degner, J.F.; Blischak, J.D.; Roux, J.; Pritchard, J.K.; Gilad, Y. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 2014, 10, e1004663. [Google Scholar] [CrossRef] [PubMed]
- Schalkwyk, L.C.; Meaburn, E.L.; Smith, R.; Dempster, E.L.; Jeffries, A.R.; Davies, M.N.; Plomin, R.; Mill, J. Allelic skewing of DNA methylation is widespread across the genome. Am. J. Hum. Genet. 2010, 86, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Arcelus, M.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Ongen, H.; Yurovsky, A.; Bryois, J.; Giger, T.; Romano, L.; Planchon, A.; et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife 2013, 2, e00523. [Google Scholar] [PubMed]
- Zhang, D.; Cheng, L.; Badner, J.A.; Chen, C.; Chen, Q.; Luo, W.; Craig, D.W.; Redman, M.; Gershon, E.S.; Liu, C. Genetic control of individual differences in gene-specific methylation in human brain. Am. J. Hum. Genet. 2010, 86, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar]
- Heinzen, E.L.; Ge, D.; Cronin, K.D.; Maia, J.M.; Shianna, K.V.; Gabriel, W.N.; Welsh-Bohmer, K.A.; Hulette, C.M.; Denny, T.N.; Goldstein, D.B. Tissue-specific genetic control of splicing: Implications for the study of complex traits. PLoS Biol. 2008, 6, e1. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.; Grundberg, E.; Koka, V.; Ge, B.; Lam, K.C.L.; Dias, C.; Kindmark, A.; Mallmin, H.; Ljunggren, O.; Rivadeneira, F.; et al. Tissue effect on genetic control of transcript isoform variation. PLoS Genet. 2009, 5, e1000608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Arcelus, M.; Ongen, H.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Yurovsky, A.; Bryois, J.; Padioleau, I.; Romano, L.; Planchon, A.; et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015, 11, e1004958. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Badner, J.A.; Cheng, L.; Zhang, C.; Zhang, D.; Cox, N.J.; Gershon, E.S.; Kelsoe, J.R.; Greenwood, T.A.; Nievergelt, C.M.; et al. Enrichment of cis-regulatory gene expression SNPs and methylation quantitative trait loci among bipolar disorder susceptibility variants. Mol. Psychiatry 2013, 18, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [Green Version]
- Liu, J.; Li, J.; Ren, Y.; Liu, P. DLG5 in cell polarity maintenance and cancer development. Int. J. Biol. Sci. 2014, 10, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Elkabetz, Y.; Studer, L. Human ESC-derived neural rosettes and neural stem cell progression. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Numata, S.; Ye, T.; Herman, M.; Lipska, B.K. DNA methylation changes in the postmortem dorsolateral prefrontal cortex of patients with schizophrenia. Front. Genet. 2014, 5, 280. [Google Scholar] [CrossRef] [PubMed]
- McRae, A.F.; Powell, J.E.; Henders, A.K.; Bowdler, L.; Hemani, G.; Shah, S.; Painter, J.N.; Martin, N.G.; Visscher, P.M.; Montgomery, G.W. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 2014, 15, R73. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Juraeva, D.; Haenisch, B.; Zapatka, M.; Frank, J.; GROUP Investigators; PSYCH-GEMS SCZ Working Group; Witt, S.H.; Mühleisen, T.W.; Treutlein, J.; Strohmaier, J.; et al. Integrated pathway-based approach identifies association between genomic regions at CTCF and CACNB2 and schizophrenia. PLoS Genet. 2014, 10, e1004345. [Google Scholar] [CrossRef] [PubMed]
- Guintivano, J.; Aryee, M.J.; Kaminsky, Z.A. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics 2013, 8, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Farlik, M.; Sheffield, N.C.; Nuzzo, A.; Datlinger, P.; Schönegger, A.; Klughammer, J.; Bock, C. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015, 10, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.-X.; Zhang, L.; Kim, A.; Li, X.; Dai, Q.; Shen, Y.; Park, B.; et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012, 149, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Qiu, R.; Wu, X.; Li, A.X.; Zhang, H.; Wang, J.; Jui, J.; Jin, S.-G.; Jiang, Y.; Pfeifer, G.P.; et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in mammalian neurogenesis. Cell Rep. 2013, 3, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Li, X.; Yan, L.; Tan, Y.; Li, R.; Zhao, Y.; Wang, Y.; Xie, J.; Zhang, Y.; Song, C.; et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014, 15, R49. [Google Scholar] [CrossRef] [PubMed]
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.D.; Wallace, C.; Plagnol, V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef] [PubMed]
- Zeltner, N.; Studer, L. Pluripotent stem cell-based disease modeling: Current hurdles and future promise. Curr. Opin. Cell Biol. 2015, 37, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Brennand, K.; Savas, J.N.; Kim, Y.; Tran, N.; Simone, A.; Hashimoto-Torii, K.; Beaumont, K.G.; Kim, H.J.; Topol, A.; Ladran, I.; et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatr. 2015, 20, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Bräuninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.-D.; Göke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.-P.; Lokman, H.; et al. Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Arloth, J.; Bogdan, R.; Weber, P.; Frishman, G.; Menke, A.; Wagner, K.V.; Balsevich, G.; Schmidt, M.V.; Karbalai, N.; Czamara, D.; et al. Genetic differences in the immediate transcriptome response to stress predict risk-related brain function and psychiatric disorders. Neuron 2015, 86, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Zovkic, I.B.; Guzman-Karlsson, M.C.; Sweatt, J.D. Epigenetic regulation of memory formation and maintenance. Learn. Mem. 2013, 20, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Baker-Andresen, D.; Ratnu, V.S.; Bredy, T.W. Dynamic DNA methylation: A prime candidate for genomic metaplasticity and behavioral adaptation. Trends Neurosci. 2013, 36, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murgatroyd, C.; Wu, Y.; Bockmühl, Y.; Spengler, D. Genes learn from stress: How infantile trauma programs us for depression. Epigenetics 2010, 5, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, D.R. From neuropathology to neurodevelopment. Lancet 1995, 346, 552–557. [Google Scholar] [CrossRef]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, A.; Ziller, M.; Spengler, D. The Future is The Past: Methylation QTLs in Schizophrenia. Genes 2016, 7, 104. https://doi.org/10.3390/genes7120104
Hoffmann A, Ziller M, Spengler D. The Future is The Past: Methylation QTLs in Schizophrenia. Genes. 2016; 7(12):104. https://doi.org/10.3390/genes7120104
Chicago/Turabian StyleHoffmann, Anke, Michael Ziller, and Dietmar Spengler. 2016. "The Future is The Past: Methylation QTLs in Schizophrenia" Genes 7, no. 12: 104. https://doi.org/10.3390/genes7120104
APA StyleHoffmann, A., Ziller, M., & Spengler, D. (2016). The Future is The Past: Methylation QTLs in Schizophrenia. Genes, 7(12), 104. https://doi.org/10.3390/genes7120104