
Electroporation of DNA into Physarum polycephalum Mitochondria: Effects on Transcription and RNA Editing in Isolated Organelles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNAs Used for Electroporation
2.2. Mitochondrial Isolation
2.3. Electroporation
2.4. Microscopy
2.5. Transcription, RT-PCR, and PCR
2.6. Dot Blot Experiments
2.7. RNA Editing Assay
3. Results
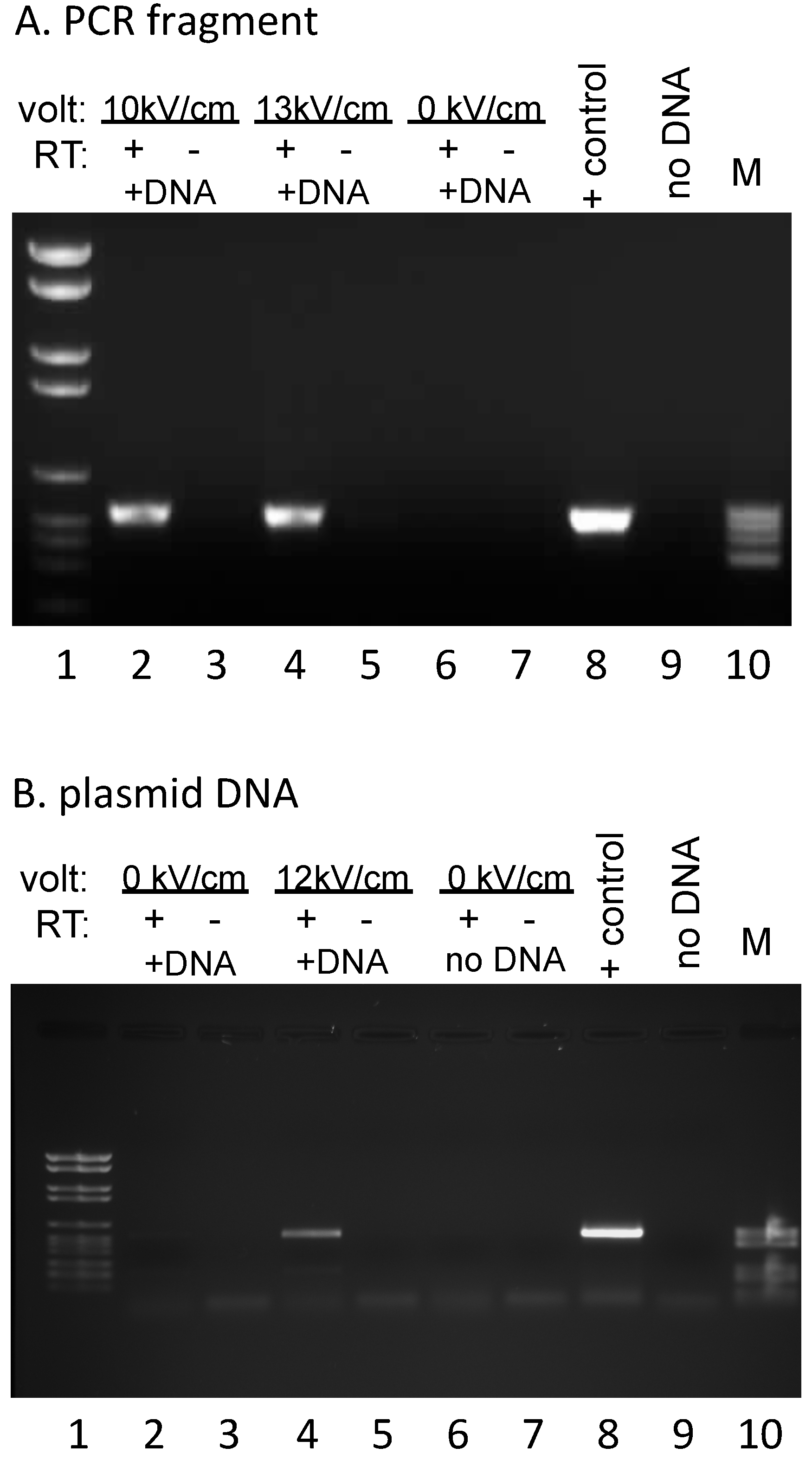
3.1. Efficient DNA Uptake upon Electroporation of Isolated P. polycephalum Mitochondria
3.2. DNA Introduced into Mitochondria is Transcribed
3.3. Transcripts Derived from DNA Introduced into Mitochondria Are Not Edited
3.4. Electroporation Does Not Appreciatively Alter the Level of Transcription from Endogenous Genes
3.5. Insertional Editing Occurs within Transcripts from Endogenous Genes after Electroporation
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Knoop, V. When you can’t trust the DNA: RNA editing changes transcript sequences. Cell. Mol. Life Sci. 2011, 68, 567–586. [Google Scholar] [CrossRef] [PubMed]
- Gott, J.M.; Emeson, R.B. Functions and mechanisms of RNA editing. Annu. Rev. Genet. 2000, 34, 499–531. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.; Zehrmann, A.; Verbitskiy, D.; Hartel, B.; Brennicke, A. RNA editing in plants and its evolution. Annu. Rev. Genet. 2013, 47, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.J.; Norman, J.E.; Schnare, M.N.; Gray, M.W.; Keeling, P.J.; Waller, R.F. Broad genomic and transcriptional analysis reveals a highly derived genome in dinoflagellate mitochondria. BMC Biol. 2007, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhang, H.; Spencer, D.F.; Norman, J.E.; Gray, M.W. Widespread and extensive editing of mitochondrial mRNAs in dinoflagellates. J. Mol. Biol. 2002, 320, 727–739. [Google Scholar] [CrossRef]
- Simpson, L.; Wang, S.H.; Thiemann, O.H.; Alfonzo, J.D.; Maslov, D.A.; Avila, H.A. U-insertion/deletion edited sequence database. Nucleic Acids Res. 1998, 26, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Bundschuh, R.; Altmuller, J.; Becker, C.; Nurnberg, P.; Gott, J.M. Complete characterization of the edited transcriptome of the mitochondrion of Physarum polycephalum using deep sequencing of RNA. Nucleic Acids Res. 2011, 39, 6044–6055. [Google Scholar] [CrossRef] [PubMed]
- Gott, J.M. Mechanisms and functions of RNA editing in Physarum polycephalum. In RNA Editing: Current Research and Future Trends; Mass, S., Ed.; Horizon Press: Norwich, UK, 2013; pp. 17–40. [Google Scholar]
- Gott, J.M.; Parimi, N.; Bundschuh, R. Discovery of new genes and deletion editing in Physarum mitochondria enabled by a novel algorithm for finding edited mRNAs. Nucleic Acids Res. 2005, 33, 5063–5072. [Google Scholar] [CrossRef] [PubMed]
- Gott, J.M.; Somerlot, B.H.; Gray, M.W. Two forms of RNA editing are required for tRNA maturation in Physarum mitochondria. RNA 2010, 16, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Gott, J.M.; Visomirski, L.M.; Hunter, J.L. Substitutional and insertional RNA editing of the cytochrome c oxidase subunit 1 mRNA of Physarum polycephalum. J. Biol. Chem. 1993, 268, 25483–25486. [Google Scholar] [PubMed]
- Antes, T.; Costandy, H.; Mahendran, R.; Spottswood, M.; Miller, D. Insertional editing of mitochondrial tRNAs of Physarum polycephalum and Didymium nigripes. Mol. Cell. Biol. 1998, 18, 7521–7527. [Google Scholar] [CrossRef] [PubMed]
- Bullerwell, C.E.; Burger, G.; Gott, J.M.; Kourennaia, O.; Schnare, M.N.; Gray, M.W. Abundant 5s rRNA-like transcripts encoded by the mitochondrial genome in amoebozoa. Eukaryot. Cell 2010, 9, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Mahendran, R.; Spottswood, M.R.; Miller, D.L. RNA editing by cytidine insertion in mitochondria of Physarum polycephalum. Nature 1991, 349, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Mahendran, R.; Spottswood, M.S.; Ghate, A.; Ling, M.L.; Jeng, K.; Miller, D.L. Editing of the mitochondrial small subunit rRNA in Physarum polycephalum. EMBO J. 1994, 13, 232–240. [Google Scholar] [PubMed]
- Wang, S.S.; Mahendran, R.; Miller, D.L. Editing of cytochrome b mRNA in Physarum mitochondria. J. Biol. Chem. 1999, 274, 2725–2731. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Visomirski-Robic, L.M.; Gott, J.M. Non-templated addition of nucleotides to the 3′ end of nascent RNA during RNA editing in Physarum. EMBO J. 2001, 20, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Gott, J.M. Transcription and RNA editing in a soluble in vitro system from Physarum mitochondria. Nucleic Acids Res. 2000, 28, 3695–3701. [Google Scholar] [CrossRef] [PubMed]
- Visomirski-Robic, L.M.; Gott, J.M. Accurate and efficient insertional RNA editing in isolated Physarum mitochondria. RNA 1995, 1, 681–691. [Google Scholar] [PubMed]
- Rhee, A.C.; Somerlot, B.H.; Parimi, N.; Gott, J.M. Distinct roles for sequences upstream of and downstream from Physarum editing sites. RNA 2009, 15, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Collombet, J.M.; Wheeler, V.C.; Vogel, F.; Coutelle, C. Introduction of plasmid DNA into isolated mitochondria by electroporation. A novel approach toward gene correction for mitochondrial disorders. J. Biol. Chem. 1997, 272, 5342–5347. [Google Scholar] [CrossRef] [PubMed]
- Castandet, B.; Araya, A. The RNA editing pattern of cox2 mRNA is affected by point mutations in plant mitochondria. PLoS ONE 2011, 6, e20867. [Google Scholar] [CrossRef] [PubMed]
- Farre, J.C.; Araya, A. Gene expression in isolated plant mitochondria: High fidelity of transcription, splicing and editing of a transgene product in electroporated organelles. Nucleic Acids Res. 2001, 29, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Farre, J.C.; Araya, A. RNA splicing in higher plant mitochondria: Determination of functional elements in group II intron from a chimeric cox II gene in electroporated wheat mitochondria. Plant J. 2002, 29, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Farre, J.C.; Leon, G.; Jordana, X.; Araya, A. Cis recognition elements in plant mitochondrion RNA editing. Mol. Cell. Biol. 2001, 21, 6731–6737. [Google Scholar] [CrossRef] [PubMed]
- Choury, D.; Farre, J.C.; Jordana, X.; Araya, A. Different patterns in the recognition of editing sites in plant mitochondria. Nucleic Acids Res. 2004, 32, 6397–6406. [Google Scholar] [CrossRef] [PubMed]
- Farre, J.C.; Aknin, C.; Araya, A.; Castandet, B. RNA editing in mitochondrial trans-introns is required for splicing. PLoS ONE 2012, 7, e52644. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, M.; Bolle, N.; Kempken, F. Mitochondrial electroporation and in organello RNA editing of chimeric atp6 transcripts. Mol. Genet. Genom. 2005, 273, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, M.; Kempken, F. Electroporation of isolated higher-plant mitochondria: Transcripts of an introduced cox2 gene, but not an atp6 gene, are edited in organello. Mol. Genet. Genom. 2003, 269, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Castandet, B.; Choury, D.; Begu, D.; Jordana, X.; Araya, A. Intron RNA editing is essential for splicing in plant mitochondria. Nucleic Acids Res. 2010, 38, 7112–7121. [Google Scholar] [CrossRef] [PubMed]
- Choury, D.; Farre, J.C.; Jordana, X.; Araya, A. Gene expression studies in isolated mitochondria: Solanum tuberosum rps10 is recognized by cognate potato but not by the transcription, splicing and editing machinery of wheat mitochondria. Nucleic Acids Res. 2005, 33, 7058–7065. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.W.; Baldwin, H.H. Methods of culture for plasmodial myxomycetes. In Methods in Cell Physiology; Prescott, D.M., Ed.; Academic Press: New York, NY, USA, 1964; Volume 1, pp. 9–41. [Google Scholar]
- Mohberg, J. Preparation of spherules. In Cell Biology of Physarum and Didymium: Differentiation, Metabolism, and Methodology; Aldruch, H.C., Daniel, J.W., Eds.; Academic Press: New York, NY, USA, 1982; Volume II, pp. 241–243. [Google Scholar]
- Byrne, E.M.; Visomirski-Robic, L.; Cheng, Y.W.; Rhee, A.C.; Gott, J.M. RNA editing in Physarum mitochondria: Assays and biochemical approaches. Methods Enzymol. 2007, 424, 143–172. [Google Scholar] [PubMed]
- Visomirski-Robic, L.M.; Gott, J.M. Insertional editing in isolated Physarum mitochondria is linked to RNA synthesis. RNA 1997, 3, 821–837. [Google Scholar] [PubMed]
- Farre, J.C.; Choury, D.; Araya, A. In organello gene expression and RNA editing studies by electroporation-mediated transformation of isolated plant mitochondria. Methods Enzymol. 2007, 424, 483–500. [Google Scholar] [PubMed]
- Byrne, E.M.; Gott, J.M. Unexpectedly complex editing patterns at dinucleotide insertion sites in Physarum mitochondria. Mol. Cell. Biol. 2004, 24, 7821–7828. [Google Scholar] [CrossRef] [PubMed]
- Visomirski-Robic, L.M.; Gott, J.M. Insertional editing of nascent mitochondrial RNAs in Physarum. Proc. Natl. Acad. Sci. USA 1997, 94, 4324–4329. [Google Scholar] [CrossRef] [PubMed]
- Byrne, E.M.; Gott, J.M. Cotranscriptional editing of Physarum mitochondrial RNA requires local features of the native template. RNA 2002, 8, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Byrne, E.M.; Stout, A.; Gott, J.M. Editing site recognition and nucleotide insertion are separable processes in Physarum mitochondria. EMBO J. 2002, 21, 6154–6161. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gott, J.M.; Naegele, G.M.; Howell, S.J. Electroporation of DNA into Physarum polycephalum Mitochondria: Effects on Transcription and RNA Editing in Isolated Organelles. Genes 2016, 7, 128. https://doi.org/10.3390/genes7120128
Gott JM, Naegele GM, Howell SJ. Electroporation of DNA into Physarum polycephalum Mitochondria: Effects on Transcription and RNA Editing in Isolated Organelles. Genes. 2016; 7(12):128. https://doi.org/10.3390/genes7120128
Chicago/Turabian StyleGott, Jonatha M., Gregory M. Naegele, and Scott J. Howell. 2016. "Electroporation of DNA into Physarum polycephalum Mitochondria: Effects on Transcription and RNA Editing in Isolated Organelles" Genes 7, no. 12: 128. https://doi.org/10.3390/genes7120128
APA StyleGott, J. M., Naegele, G. M., & Howell, S. J. (2016). Electroporation of DNA into Physarum polycephalum Mitochondria: Effects on Transcription and RNA Editing in Isolated Organelles. Genes, 7(12), 128. https://doi.org/10.3390/genes7120128