
Tissue Non-Specific Genes and Pathways Associated with Diabetes: An Expression Meta-Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Datasets
2.2. Gene Expression Association Test and Meta-Analysis
2.3. Pathway Expression Association Test and Meta-Analysis
2.4. KEGG Pathway Mapping Analysis
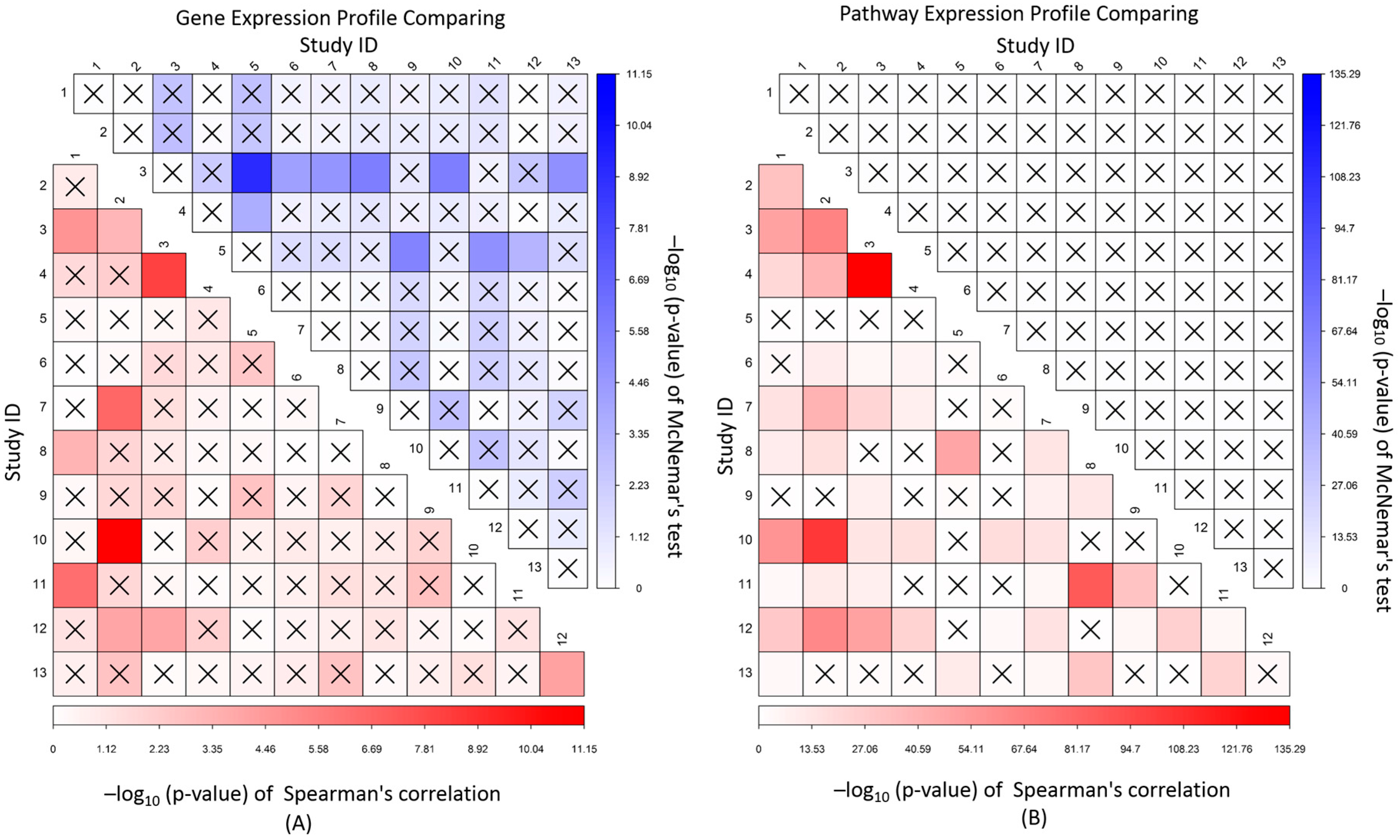
2.5. Correlation and Independent Analysis of Expression Association Profile between Studies
3. Results
3.1. Characteristics of the Gene Expression Datasets and Studies
3.2. Tissue Non-Specific Gene Expression Association
3.3. Tissue Non-Specific Pathway Expression Association
3.4. Mapped KEGG Pathways for the Identified Gene Sets
3.5. Correlation and Independence of Gene and Pathway Expression Associations
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alberti, K.G.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a who consultation. Diabet. Med. J. Br. Diabet. Assoc. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, C.M.; Hojlund, K.; Hansen, L.; Oakeley, E.J.; Hemmings, B.; Abdallah, B.M.; Brusgaard, K.; Beck-Nielsen, H.; Gaster, M. Transcriptional profiling of myotubes from patients with type 2 diabetes: No evidence for a primary defect in oxidative phosphorylation genes. Diabetologia 2008, 51, 2068–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oostrom, O.; de Kleijn, D.P.; Fledderus, J.O.; Pescatori, M.; Stubbs, A.; Tuinenburg, A.; Lim, S.K.; Verhaar, M.C. Folic acid supplementation normalizes the endothelial progenitor cell transcriptome of patients with type 1 diabetes: A case-control pilot study. Cardiovasc. Diabetol. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cangemi, C.; Skov, V.; Poulsen, M.K.; Funder, J.; Twal, W.O.; Gall, M.A.; Hjortdal, V.; Jespersen, M.L.; Kruse, T.A.; Aagard, J.; et al. Fibulin-1 is a marker for arterial extracellular matrix alterations in type 2 diabetes. Clin. Chem. 2011, 57, 1556–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skov, V.; Knudsen, S.; Olesen, M.; Hansen, M.L.; Rasmussen, L.M. Global gene expression profiling displays a network of dysregulated genes in non-atherosclerotic arterial tissue from patients with type 2 diabetes. Cardiovasc. Diabetol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein p, causes insulin resistance. Cell Metab. 2010, 12, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Pihlajamaki, J.; Boes, T.; Kim, E.Y.; Dearie, F.; Kim, B.W.; Schroeder, J.; Mun, E.; Nasser, I.; Park, P.J.; Bianco, A.C.; et al. Thyroid hormone-related regulation of gene expression in human fatty liver. J. Clin. Endocrinol. Metab. 2009, 94, 3521–3529. [Google Scholar] [CrossRef] [PubMed]
- Kaizer, E.C.; Glaser, C.L.; Chaussabel, D.; Banchereau, J.; Pascual, V.; White, P.C. Gene expression in peripheral blood mononuclear cells from children with diabetes. J. Clin. Endocrinol. Metab. 2007, 92, 3705–3711. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; et al. Ncbi geo: Archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2011, 39, D1005–D1010. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. Geoquery: A bridge between the gene expression omnibus (GEO) and bioconductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Sigdel, T.K.; Li, L.; Kambham, N.; Dudley, J.T.; Hsieh, S.C.; Klassen, R.B.; Chen, A.; Caohuu, T.; Morgan, A.A.; et al. Differentially expressed rna from public microarray data identifies serum protein biomarkers for cross-organ transplant rejection and other conditions. PLoS Comput. Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Bolstad bm. Preprocesscore: A Collection of Pre-processing Functions. R Package Version 1.36.0. 2016. Available online: https://github.Com/bmbolstad/preprocesscore (accessed on 1 March 2006).
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Li, L.; Liu, S.; Jiang, F.; Griswold, M.; Mosley, T. The uniform-score gene set analysis for identifying common pathways associated with different diabetes traits. BMC Genom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A. A description of the molecular signatures database (MSigDb) web site. Methods Mol. Biol. 2014, 1150, 153–160. [Google Scholar] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. Kegg for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Li, L.; Jiang, F.; Simino, J.; Griswold, M.; Mosley, T.; Liu, S. snpGeneSets: An R package for genome-wide study annotation. G3 2016, 6, 4087–4095. [Google Scholar] [PubMed]
- Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 2010, 36, 1–48. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. Kegg for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef] [PubMed]
- Karolina, D.S.; Armugam, A.; Tavintharan, S.; Wong, M.T.; Lim, S.C.; Sum, C.F.; Jeyaseelan, K. Microrna 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS ONE 2011, 6, e22839. [Google Scholar] [CrossRef]
- Marselli, L.; Thorne, J.; Dahiya, S.; Sgroi, D.C.; Sharma, A.; Bonner-Weir, S.; Marchetti, P.; Weir, G.C. Gene expression profiles of beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS ONE 2010, 5, e11499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, V.; Raimondi, C.; Somanath, S.; Bugliani, M.; Loder, M.K.; Edling, C.E.; Divecha, N.; da Silva-Xavier, G.; Marselli, L.; Persaud, S.J.; et al. Class ii phosphoinositide 3-kinase regulates exocytosis of insulin granules in pancreatic beta cells. J. Biol. Chem. 2011, 286, 4216–4225. [Google Scholar] [CrossRef] [PubMed]
- Taneera, J.; Lang, S.; Sharma, A.; Fadista, J.; Zhou, Y.; Ahlqvist, E.; Jonsson, A.; Lyssenko, V.; Vikman, P.; Hansson, O.; et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012, 16, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Van Tienen, F.H.; Praet, S.F.; de Feyter, H.M.; van den Broek, N.M.; Lindsey, P.J.; Schoonderwoerd, K.G.; de Coo, I.F.; Nicolay, K.; Prompers, J.J.; Smeets, H.J.; et al. Physical activity is the key determinant of skeletal muscle mitochondrial function in type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 3261–3269. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Goldfine, A.B.; Boes, T.; Henry, R.R.; Ciaraldi, T.P.; Kim, E.Y.; Emecan, M.; Fitzpatrick, C.; Sen, A.; Shah, A.; et al. Increased srf transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. J. Clin. Investig. 2011, 121, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Pratley, R.E.; Tokraks, S.; Bogardus, C.; Permana, P.A. Microarray profiling of skeletal muscle tissues from equally obese, non-diabetic insulin-sensitive and insulin-resistant pima indians. Diabetologia 2002, 45, 1584–1593. [Google Scholar] [PubMed]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. Txnip regulates peripheral glucose metabolism in humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coletta, D.K.; Balas, B.; Chavez, A.O.; Baig, M.; Abdul-Ghani, M.; Kashyap, S.R.; Folli, F.; Tripathy, D.; Mandarino, L.J.; Cornell, J.E.; et al. Effect of acute physiological hyperinsulinemia on gene expression in human skeletal muscle in vivo. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E910–E917. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, J.; Cui, X.; Maianu, L.; Rhees, B.; Rosinski, J.; So, W.V.; Willi, S.M.; Osier, M.V.; Hill, H.S.; et al. The effect of insulin on expression of genes and biochemical pathways in human skeletal muscle. Endocrine 2007, 31, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Patki, A.; Lara-Castro, C.; Cui, X.; Zhang, K.; Walton, R.G.; Osier, M.V.; Gadbury, G.L.; Allison, D.B.; Martin, M.; et al. Genes and biochemical pathways in human skeletal muscle affecting resting energy expenditure and fuel partitioning. J. Appl. Physiol. 2011, 110, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Hardy, O.T.; Perugini, R.A.; Nicoloro, S.M.; Gallagher-Dorval, K.; Puri, V.; Straubhaar, J.; Czech, M.P. Body mass index-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surg. Obes. Relat. Dis. 2011, 7, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Stenbit, A.E.; Tsao, T.S.; Li, J.; Burcelin, R.; Geenen, D.L.; Factor, S.M.; Houseknecht, K.; Katz, E.B.; Charron, M.J. Glut4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat. Med. 1997, 3, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Peroni, O.; Kim, J.K.; Kim, Y.B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-selective targeting of the glut4 gene impairs insulin action in muscle and liver. Nature 2001, 409, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 2000, 6, 924–928. [Google Scholar] [PubMed]
- Zhang, M.; Robitaille, M.; Showalter, A.D.; Huang, X.; Liu, Y.; Bhattacharjee, A.; Willard, F.S.; Han, J.; Froese, S.; Wei, L.; et al. Progesterone receptor membrane component 1 is a functional part of the glucagon-like peptide-1 (GLP-1) receptor complex in pancreatic beta cells. Mol. Cell Proteom. 2014, 13, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Senniappan, S.; Shanti, B.; James, C.; Hussain, K. Hyperinsulinaemic hypoglycaemia: Genetic mechanisms, diagnosis and management. J. Inherit. Metab. Dis. 2012, 35, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, Z.H.; Ren, Z.; Qu, S.L.; Liu, M.H.; Liu, L.S.; Jiang, Z.S. Hydrogen sulfide, the next potent preventive and therapeutic agent in aging and age-associated diseases. Mol. Cell. Biol. 2013, 33, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.M.; Lee, J.H.; Yadav, H.; Kamaraju, A.K.; Liu, E.; Zhigang, D.; Vieira, A.; Kim, S.J.; Collins, H.; Matschinsky, F.; et al. Transforming growth factor-beta/smad3 signaling regulates insulin gene transcription and pancreatic islet beta-cell function. J. Biol. Chem. 2009, 284, 12246–12257. [Google Scholar] [CrossRef] [PubMed]
- Veneri, D.; Franchini, M.; Bonora, E. Imatinib and regression of type 2 diabetes. N. Engl. J. Med. 2005, 352, 1049–1050. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Bonner-Weir, S.; Li, W.C.; Ouziel-Yahalom, L.; Guo, L.; Weir, G.C.; Sharma, A. Beta-cell growth and regeneration: Replication is only part of the story. Diabetes 2010, 59, 2340–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simone, S.; Gorin, Y.; Velagapudi, C.; Abboud, H.E.; Habib, S.L. Mechanism of oxidative DNA damage in diabetes: Tuberin inactivation and downregulation of DNA repair enzyme 8-oxo-7,8-dihydro-2’-deoxyguanosine-DNA glycosylase. Diabetes 2008, 57, 2626–2636. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Watson, G.S. Insulin and neurodegenerative disease: Shared and specific mechanisms. Lancet Neurol. 2004, 3, 169–178. [Google Scholar] [CrossRef]
- Ferre, P. The biology of peroxisome proliferator-activated receptors: Relationship with lipid metabolism and insulin sensitivity. Diabetes 2004, 53, S43–S50. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study | GDS_ID | GPL_ID | Pub_ID | N_genes | Size | Contrast | Tissue |
|---|---|---|---|---|---|---|---|
| Diabetes State | |||||||
| 1 | GDS3665 | GPL2986 | 16,075 | 10 | T2D vs. control | adipose | |
| 2 | GDS3980 | GPL571 | 21926180 [6]; 22340758 [7] | 12,778 | 21 | T2D vs. control | artery |
| 3 | GDS3874 | GPL96 | 17595242 [10] | 18,552 | 117 | T1D vs. healthy and T2D vs. healthy | blood |
| GDS3875 | GPL97 | ||||||
| 4 | GDS3963 | GPL6883 | 21829658 [23] | 17,476 | 24 | T2D vs. impaired fasting glucose vs. control | blood |
| 5 | GDS3656 | GPL2700 | 19706161 [5] | 16,778 | 32 | T1D vs. Healthy | EPC |
| 6 | GDS3876 | GPL96 | 19549744 [9] | 12,779 | 18 | obese T2D vs. obese no T2D | liver |
| 7 | GDS3883 | GPL570 | 21035759 [8] | 20,539 | 17 | T2D vs. normal glucose tolerance | liver |
| 8 | GDS3681 | GPL8300 | 18719883 [4] | 8861 | 20 | T2D vs. control | myotube |
| 9 | GDS3782 | GPL1352 | 20644627 [24] | 20,185 | 20 | T2D vs. control | pancreas |
| 10 | GDS3882 | GPL96 | 21127054 [25] | 12,779 | 13 | T2D vs. non-diabetes | pancreas |
| 11 | GDS4337 | GPL6244 | 22768844 [26] | 17,323 | 63 | T2D vs. non-diabetes | pancreas |
| 12 | GDS3880 | GPL570 | 22802091 [27] | 20,539 | 42 | T2D vs. pre-diabetes vs. normoglycemic control | skeletal muscle |
| 13 | GDS3884 | GPL570 | 21393865 [28] | 20,539 | 50 | T2D vs. Normoglycemia with FH+ vs. Normoglycemia with FH− | skeletal muscle |
| Insulin Action | |||||||
| 1 | GDS157 | GPL80 | 12436343 [29] | 13,742 | 10 | insulin resistant vs. insulin sensitive | skeletal muscle |
| GDS158 | GPL98 | ||||||
| GDS160 | GPL99 | ||||||
| GDS161 | GPL100 | ||||||
| GDS162 | GPL101 | ||||||
| 2 | GDS2790 | GPL80 | 17472435 [30] | 12,885 | 12 | Before vs. after Hyperinsulinemic-euglycemic clamp for nondiabetes | skeletal muscle |
| GDS2791 | GPL96 | ||||||
| 3 | GDS3181 | GPL96 | 18334611 [31] | 12,779 | 36 | −60 vs. 30 vs. 240 min of Hyperinsulinemic-euglycemic clamp for nondiabetes | skeletal muscle |
| 4 | GDS3715 | GPL91 | 17709892 [32]; 21109598 [33] | 8768 | 110 | Diabetes vs. insulin sensitive vs. insulin resistant before and after Hyperinsulinemic-euglycemic clamp | skeletal muscle |
| 5 | GDS3781 | GPL570 | 20678967 [34] | 20,539 | 39; 19 | insulin sensitive vs. insulin resistant | adipose |
| GDS3962 | |||||||
| PGRMC1 | HADH | IRS1 | MPST | ||
|---|---|---|---|---|---|
| Study | GDS_ID | Gene U-Score (%) of Diabetes State | |||
| 1 | GDS3665 | 4.79 | 0.48 | 9.17 | 2.68 |
| 2 | GDS3980 | 31.31 | 14.46 | 20.64 | 3.64 |
| 3 | GDS3874/GDS3875 | 8.55 | 3.48 | 24.46 | 97.48 |
| 4 | GDS3963 | 45.03 | 48.6 | 3.01E−03 | 0.2 |
| 5 | GDS3656 | 0.52 | 3.35 | 68.95 | 89.07 |
| 6 | GDS3876 | 4.16 | 82.07 | 89.31 | 35.61 |
| 7 | GDS3883 | 97.9 | 69.24 | 15.34 | 3.85 |
| 8 | GDS3681 | 88.77 | 2.73 | 81.4 | 27.87 |
| 9 | GDS3782 | 0.34 | 7.43 | 81 | 79.18 |
| 10 | GDS3882 | 2.33 | 66.82 | 95.75 | 47.81 |
| 11 | GDS4337 | 4.66 | 0.44 | 7.54 | 37.53 |
| 12 | GDS3880 | 95.17 | 50.5 | 41.35 | 6.55 |
| 13 | GDS3884 | 96.25 | 2.97 | 48.08 | 54.24 |
| Bin_P | 1.03E−6 | 1.03E−6 | 0.14 | 2.87E−4 | |
| Study | GDS_ID | Gene U-Score (%) of Insulin Action | |||
| 1 | GDS157/GDS158/GDS160/GDS161/GDS162 | 7.48 | NA | 3.82 | 2.2 |
| 2 | GDS2790/GDS2791 | 9.59 | 10.14 | 2.5 | 3.92 |
| 3 | GDS3181 | 55.03 | 16.78 | 3.46 | 4.56 |
| 4 | GDS3715 | 30.9 | 82.89 | 25.76 | 0.86 |
| 5 | GDS3781/GDS3962 | 48.65 | 3.71 | 4.52 | 23.59 |
| Bin_P | 0.23 | 0.01 | 3.13E−7 | 3.13E−7 | |
| Joint Analysis of Combined Diabetes State and Insulin Action | |||||
| Bin_P | 6.31E−7 | 6.28E−8 | |||
| PID | GeneSet | Fixed_p | Bin_p0 | Bin_p1 |
|---|---|---|---|---|
| Diabetes Studies | ||||
| 5599 | UV response | 7.72E−17 | 4.01E−08 | 3.10E−03 |
| 4914 | chronic myelogenous leukemia | 1.45E−38 | 1.16E−09 | 1.97E−05 |
| 7922 | KLF1 targets | 3.35E−26 | 3.47E−13 | 3.10E−03 |
| 5947 | SMARCA2 targets | 1.95E−25 | 4.01E−08 | 3.10E−03 |
| 6442 | Alzheimer’s disease | 1.65E−19 | 4.01E−08 | 2.87E−04 |
| 7145 | stromal stem cells | 1.88E−15 | 1.16E−09 | 2.87E−04 |
| Insulin Response Studies | ||||
| 5599 | UV response | 1.48E−18 | 3.00E−05 | 0.023 |
| 4914 | chronic myelogenous leukemia | 4.27E−08 | 3.00E−05 | 0.023 |
| 7922 | KLF1 targets | 4.55E−05 | 3.00E−05 | 0.023 |
| 5947 | SMARCA2 targets | 1.12E−04 | 0.023 | 0.023 |
| 6442 | Alzheimer’s disease | 0.042 | 1.16E−03 | 0.23 |
| 7145 | stromal stem cells | 2.14E−08 | 1.11E−03 | 0.023 |
| Joint Analysis | ||||
| 5599 | UV response | 9.46E−32 | 1.12E−10 | 1.55E−03 |
| 4914 | chronic myelogenous leukemia | 3.91E−44 | 3.41E−12 | 1.52E−05 |
| 7922 | KLF1 targets | 4.72E−29 | 1.54E−15 | 1.55E−03 |
| 5947 | SMARCA2 targets | 1.11E−27 | 6.28E−08 | 1.55E−03 |
| 6442 | Alzheimer’s disease | 1.84E−18 | 2.95E−09 | 1.55E−03 |
| 7145 | stromal stem cells | 2.93E−22 | 1.12E−10 | 1.72E−04 |
| Gene Set | KEGG Pathway | Size | Gene | Effect | SE | p | adj_p |
|---|---|---|---|---|---|---|---|
| UV response | TGF-beta signaling pathway | 86 | 22 | 0.20 | 0.03 | 1.28E−09 | <0.001 |
| chronic myelogenous leukemia | The citrate cycle | 32 | 15 | 0.35 | 0.06 | 1.11E−06 | <0.001 |
| KLF1 targets | DNA replication | 36 | 13 | 0.24 | 0.05 | 1.54E−04 | 0.016 |
| SMARCA2 targets | Nucleotide excision repair | 44 | 6 | 0.11 | 0.02 | 4.29E−04 | 0.034 |
| Alzheimer’s disease | 1. Oxidative phosphorylation | 135 | 62 | 0.35 | 0.03 | 9.20E−27 | <0.001 |
| 2. Parkinson’s disease | 133 | 60 | 0.35 | 0.03 | 2.13E−25 | <0.001 | |
| stromal stem cells | 1. PPAR signaling pathway | 69 | 9 | 0.10 | 0.02 | 3.02E−4 | 0.029 |
| 2. p53 signaling pathway | 69 | 9 | 0.10 | 0.02 | 3.02E−4 | 0.029 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, H.; Li, L.; Liu, S.; Jiang, F.; Griswold, M.; Mosley, T. Tissue Non-Specific Genes and Pathways Associated with Diabetes: An Expression Meta-Analysis. Genes 2017, 8, 44. https://doi.org/10.3390/genes8010044
Mei H, Li L, Liu S, Jiang F, Griswold M, Mosley T. Tissue Non-Specific Genes and Pathways Associated with Diabetes: An Expression Meta-Analysis. Genes. 2017; 8(1):44. https://doi.org/10.3390/genes8010044
Chicago/Turabian StyleMei, Hao, Lianna Li, Shijian Liu, Fan Jiang, Michael Griswold, and Thomas Mosley. 2017. "Tissue Non-Specific Genes and Pathways Associated with Diabetes: An Expression Meta-Analysis" Genes 8, no. 1: 44. https://doi.org/10.3390/genes8010044
APA StyleMei, H., Li, L., Liu, S., Jiang, F., Griswold, M., & Mosley, T. (2017). Tissue Non-Specific Genes and Pathways Associated with Diabetes: An Expression Meta-Analysis. Genes, 8(1), 44. https://doi.org/10.3390/genes8010044