
Alternative Splicing in Breast Cancer and the Potential Development of Therapeutic Tools



{kind=link}
Abstract
:1. Introduction
2. Current BrCa Therapeutics
3. Alternative Splicing in Cancer
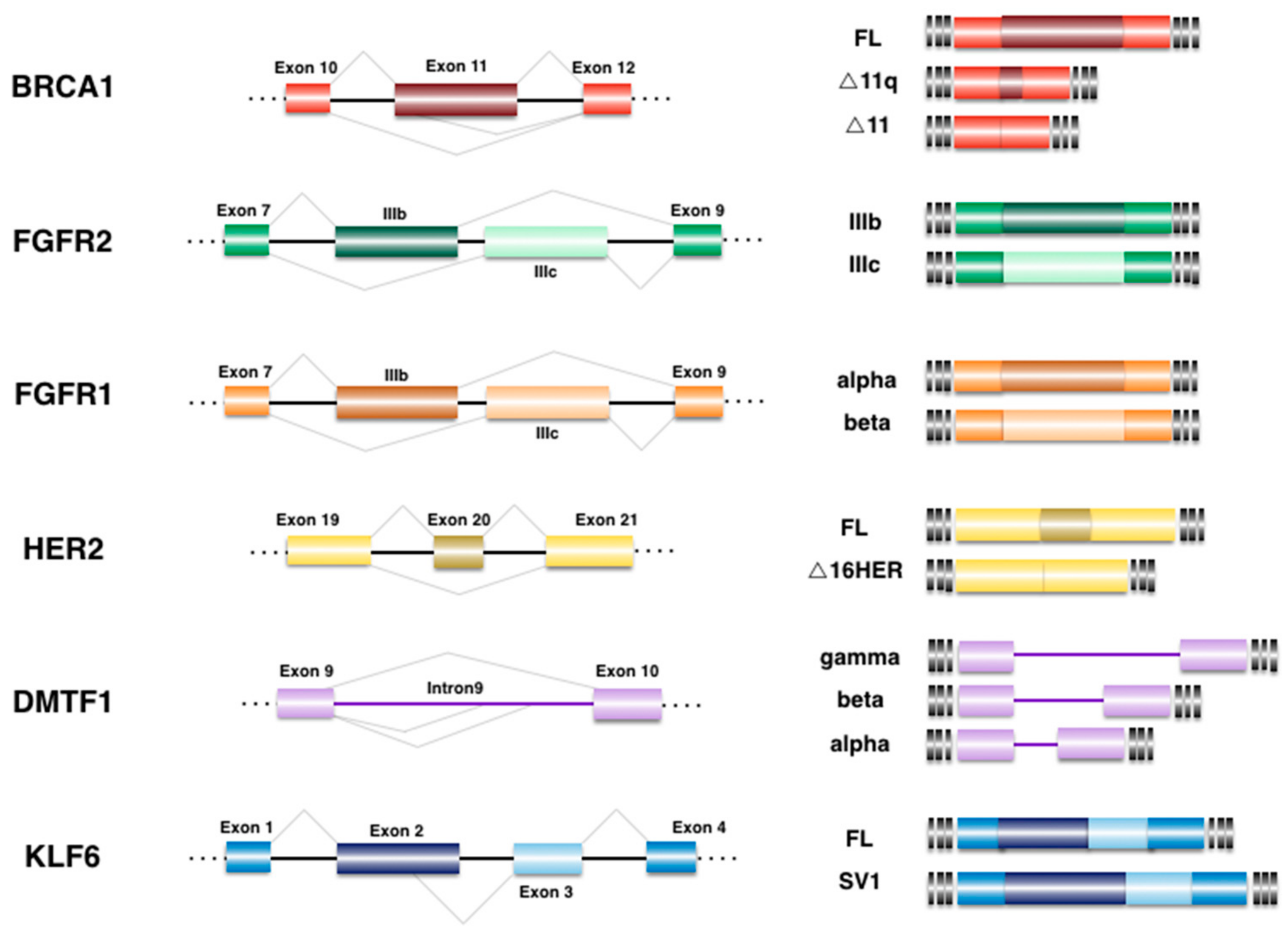
4. Alternative Splicing Events Associated to BrCa
4.1. Breast Cancer 1 (BRCA1)
4.2. Cyclin D-Binding myb-like Transcription Factor 1 (DMTF1)
4.3. Epidermal Growth Factor Receptor 2 (HER2)
4.4. Fibroblast Growth Factor Receptor (FGFR)
4.5. Krüppel-like Zinc Finger Factor 6 (KLF6)
4.6. Survivin
4.7. TP53
5. Prognostic Value of AS Variants in Breast Cancer
6. Modification of Splicing Events as a Therapeutic Approach
7. AS events and Chemotherapeutic Response
8. AS and Endocrine Therapy in BrCa
9. Conclusions
Conflicts of Interest
References
- Anaya-Ruiz, M.; Vallejo-Ruiz, V.; Flores-Mendoza, L.; Perez-Santos, M. Female Breast Cancer Incidence and Mortality in Mexico. Asian Pac. J. Cancer Prev. 2014, 15, 1477–1479. [Google Scholar] [CrossRef] [PubMed]
- Dogan, N.; Toprak, D. Female breast cancer mortality rates in Turkey. Asian Pac. J. Cancer Prev. 2014, 15, 7569–7573. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.J.; Au, W.W.; Wu, K.S.; Chen, L.X.; Lin, K. Mortality characteristics and prediction of female breast cancer in China from 1991 to 2011. Asian Pac. J. Cancer Prev. 2014, 15, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Malvezzi, M.; Bertuccio, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2014. Ann. Oncol. 2014, 25, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Youlden, D.R.; Cramb, S.M.; Yip, C.H.; Baade, P.D. Incidence and mortality of female breast cancer in the Asia-Pacific region. Cancer Biol. Med. 2014, 11, 101–115. [Google Scholar] [PubMed]
- Taghavi, A.; Fazeli, Z.; Vahedi, M.; Baghestani, A.R.; Pourhoseingholi, A.; Barzegar, F.; Pourhoseingholi, M.A. Increased trend of breast cancer mortality in Iran. Asian Pac. J. Cancer Prev. 2012, 13, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Shaukat, U.; Ismail, M.; Mehmood, N. Epidemiology, major risk factors and genetic predisposition for breast cancer in the Pakistani population. Asian Pac. J. Cancer Prev. 2013, 14, 5625–5629. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, O.; Bray, F.; Coleman, M.P.; Vanderpuye, V.; Eniu, A.; Kotha, S.R.; Sarker, M.; Huong, T.T.; Allemani, C.; Dvaladze, A.; et al. The global burden of women’s cancers: A grand challenge in global health. Lancet 2017, 389, 847–860. [Google Scholar] [CrossRef]
- Leong, S.P.; Shen, Z.Z.; Liu, T.J.; Agarwal, G.; Tajima, T.; Paik, N.S.; Sandelin, K.; Derossis, A.; Cody, H.; Foulkes, W.D. Is breast cancer the same disease in Asian and Western countries? World J. Surg. 2010, 34, 2308–2324. [Google Scholar] [CrossRef] [PubMed]
- Kohler, R.E.; Goyal, R.K.; Lich, K.H.; Domino, M.E.; Wheeler, S.B. Association between medical home enrollment and health care utilization and costs among breast cancer patients in a state Medicaid program. Cancer 2015, 121, 3975–3981. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Rentería, E.; Conway, D.I.; Bray, F.; Van Ourti, T.; Soerjomataram, I. Inequalities in cancer incidence and mortality across medium to highly developed countries in the twenty-first century. Cancer Causes Control 2016, 27, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, B.; Shimokata, T.; Honda, K.; Tsukuura, H.; Ando, Y. Should low-income countries invest in breast cancer screening? Cancer Causes Control 2016, 27, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac. J. Cancer Prev. 2015, 17, 43–46. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Neo, S.Y.; McShane, L.M.; Korn, E.L.; Long, P.M.; Jazaeri, A.; Martiat, P.; Fox, S.B.; Harris, A.L.; Liu, E.T. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl. Acad. Sci. USA 2003, 100, 10393–10398. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.; Perou, C.; Livasy, C.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.H.; Edmiston, S.; et al. Race, breast cancer sub-types, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.; Dees, E.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative para-dox: Primary tumor chemosensitivity of breast cancer sub-types. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Phipps, A.I.; Malone, K.E.; Porter, P.L.; Daling, J.R.; Li, C.I. Reproductive and hormonal risk factors for postmenopausal luminal, HER-2-overexpressing, and triple-negative breast cancer. Cancer 2008, 113, 1521–1526. [Google Scholar] [CrossRef] [PubMed]
- Holen, I.; Speirs, V.; Morrissey, B.; Blyth, K. In vivo models in breast cancer research: Progress, challenges and future directions. Dis. Model. Mech. 2017, 10, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- O’Shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Chou Koo, I.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Steger, G. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor- positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [PubMed]
- Dhankhar, R.; Vyas, S.P.; Jain, A.K.; Arora, S.; Rath, G.; Goyal, A.K. Advances in novel drug delivery strategies for breast cancer therapy. Artif. Cells Blood Subst. Immobil. Biotechnol. 2010, 38, 230–249. [Google Scholar] [CrossRef] [PubMed]
- Wuerstlein, R.; Harbeck, N. Neoadjuvant therapy for HER2-positive breast cancer. Rev. Recent Clin. Trials 2017. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Ramalho, S.; Gonçalves, R.; Barrios, C.H.; Graudenz, M.S.; Bines, J. Multidisciplinary Approach to Neoadjuvant Endocrine Therapy in Breast Cancer: A Comprehensive Review. Rev. Bras. Ginecol. Obstet. 2016, 38, 615–622. [Google Scholar] [PubMed]
- Castrellon, A.B.; Pidhorecky, I.; Valero, V.; Raez, L.E. The role of carboplatin in the neoadjuvant chemotherapy treatment of triple negative breast cancer. Oncol. Rev. 2017, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A. nab-Paclitaxel for the treatment of breast cancer: An update across treatment settings. Exp. Hematol. Oncol. 2017, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Puhalla, S.; Brufsky, A.; Davidson, N. Adjuvant endocrine therapy for premenopausal women with breast cancer. Breast 2009, 18, S122–S130. [Google Scholar] [CrossRef]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Medeiros Alencar, V.H.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013, 381, 805–816. [Google Scholar] [CrossRef]
- Yardley, D.A. Combining mTOR inhibitors with chemotherapy and other targeted therapies in advanced breast cancer: Rationale, clinical experience, and future directions. Breast Cancer 2013, 7, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Cadoo, K.A.; Gucalp, A.; Traina, T.A. Palbociclib: An evidence-based review of its potential in the treatment of breast cancer. Breast Cancer 2014, 6, 123–133. [Google Scholar] [PubMed]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef] [PubMed]
- Law, A.M.; Lim, E.; Ormandy, C.J.; Gallego-Ortega, D. The innate and adaptive infiltrating immune systems as targets for breast cancer immunotherapy. Endocr. Relat. Cancer 2017, 24, R123–R144. [Google Scholar] [CrossRef] [PubMed]
- McCrudden, C.M.; McCarthy, H.O. Current status of gene therapy for breast cancer: Progress and challenges. Appl. Clin. Genet. 2014, 7, 209. [Google Scholar] [PubMed]
- Shamsi, M.; Islamian, J.P. Breast cancer: Early diagnosis and effective treatment by drug delivery tracing. Nucl. Med. Rev. Cent. East Eur. 2017, 20, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.; Morris, D.G. Oncolytic viral therapy using reovirus. Methods Mol. Biol. 2015, 1317, 187–223. [Google Scholar] [PubMed]
- Gholami, S.; Marano, A.; Chen, N.G.; Aguilar, R.J.; Frentzen, A.; Chen, C.H.; Lou, E.; Fujisawa, S.; Eveno, C.; Belin, L.; et al. A novel vaccinia virus with dual oncolytic and anti-angiogenic therapeutic effects against triple-negative breast cancer. Breast Cancer Res. Treat. 2014, 148, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Huang, Q.; Qiu, F.; Sui, M. Non-viral delivery systems for the application in p53 cancer gene therapy. Curr. Med. Chem. 2015, 22, 4118–4136. [Google Scholar] [CrossRef] [PubMed]
- Templeton, N.S. Nonviral delivery for genomic therapy of cancer. World J. Surg. 2009, 33, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Huang, L. Non-viral nanocarriers for siRNA delivery in breast cancer. J. Control Release 2014, 190, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.B.; Yao, M.; Brummer, G.; Acevedo, D.; Alhakamy, N.; Berkland, C.; Cheng, N. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget 2016, 7, 49349. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Ma, W.H.; Ge, Y.L.; Xue, M.L.; Zhang, Z.; Zhang, J.Y.; Hou, L.; Mu, R.H. RNAi-mediated gene silencing of vascular endothelial growth factor C suppresses growth and induces apoptosis in mouse breast cancer in vitro and in vivo. Oncol. Lett. 2016, 12, 3896–3904. [Google Scholar] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar]
- Hu, X.C.; Zhang, J.; Xu, B.H.; Cai, L.; Ragaz, J.; Wang, Z.H.; Wang, B.Y.; Teng, Y.E.; Tong, Z.S.; Pan, Y.Y.; et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 436–446. [Google Scholar] [PubMed]
- Rugo, H.S.; Olopade, O.I.; DeMichele, A.; Yau, C.; van ’t Veer, L.J.; Buxton, M.B.; Hogarth, M.; Hylton, N.M.; Paoloni, M.; Perlmutter, J.; et al. Adaptive randomization of veliparib-carboplatin treatment in breast cancer. N. Engl. J. Med. 2016, 375, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Boér, K. Impact of palbociclib combinations on treatment of advanced estrogen receptor-positive/human epidermal growth factor 2-negative breast cancer. Onco Targets Ther. 2016, 9, 6119. [Google Scholar] [CrossRef] [PubMed]
- Kaczyńska, A.; Herman-Antosiewicz, A. Combination of lapatinib with isothiocyanates overcomes drug resistance and inhibits migration of HER2 positive breast cancer cells. Breast Cancer 2016, 24, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Matlin, A.J.; Clark, F.; Smith, C.W. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005, 6, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Ladomery, M. Aberrant Alternative Splicing Is Another Hallmark of Cancer. Int. J. Cell Biol. 2013, 2013, 463786. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Montiel, N.; Rosas-Murrieta, N.H.; Martínez-Contreras, R. Alternative splicing regulation: Implications in cancer diagnosis and treatment. Med. Clin. 2015, 144, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Montiel, N.; Rosas-Murrieta, N.H.; Martínez-Montiel, M.; Gaspariano-Cholula, M.P.; Martínez-Contreras, R.D. Microbial and natural metabolites that inhibit splicing: A powerful alternative for cancer treatment. BioMed Res. Int. 2016, 3681094. [Google Scholar] [CrossRef] [PubMed]
- Sebestyen, E.; Zawisza, M.; Eyras, E. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res. 2015, 43, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Silipo, M.; Gautrey, H.; Tyson-Capper, A. Deregulation of splicing factors and breast cancer development. J. Mol. Cell. Biol. 2015, 7, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Vanharanta, S.; Marney, C.B.; Shu, W.; Valiente, M.; Zou, Y.; Mele, A.; Darnell, R.B.; Massagué, J. Loss of the multifunctional RNA-binding protein RBM47 as a source of selectable metastatic traits in breast cancer. Elife 2014, 3, e02734. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Akerman, M.; Cléry, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-regulated alternative splicing in breast cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Takeda, J.-I.; Suzuki, Y.; Sakate, R.; Sato, Y.; Seki, M.; Irie, T.; Takeuchi, N.; Ueda, T.; Nakao, M.; Sugano, S.; et al. Low conservation and species-specific evolution of alternative splicing in humans and mice: Comparative genomics analysis using well-annotated full-length cDNAs. Nucleic Acids Res. 2008, 36, 6386–6395. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Schad, E.; Tompa, P.; Hegyi, H. The relationship between proteome size, structural disorder and organism complexity. Genome Biol. 2011, 12, R120. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 2014, 343, 1470–1475. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.X. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Zhang, H.B.; Peng, Y.; Le, H.; Carroll, B.; Ward, T.; Yao, J.; Farid, L.M.; Couch, F.J.; Wilson, R.B.; et al. Localization of BRCA1 and a splice variant identifies the nuclear localization signal. Mol. Cell. Biol. 1997, 17, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Cao, L.; Lim, S.C.; Le, H.; Carroll, B.; Ward, T.; Yao, J.; Farid, L.M.; Couch, F.J.; Wilson, R.B.; et al. Hyperplasia and spontaneous tumor development in the gynecologic system in mice lacking the BRCA1-Delta11 isoform. Mol. Cell. Biol. 2006, 26, 6983–6992. [Google Scholar] [CrossRef] [PubMed]
- Wiener, D.; Gajardo-Meneses, P.; Ortega-Hernández, V.; Herrera-Cares, C.; Díaz, S.; Fernández, W.; Cornejo, V.; Gamboa, J.; Tapia, T.; Alvarez, C.; et al. BRCA1 and BARD1 colocalize mainly in the cytoplasm of breast cancer tumors, and their isoforms show differential expression. Breast Cancer Res. Treat. 2015, 153, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Blok, M.J.; Whiley, P.; Santamariña, M.; Gutiérrez-Enríquez, S.; Romero, A.; Garre, P.; Becker, A.; Smith, L.D.; De Vecchi, G.; et al. Comprehensive annotation of splice junctions supports pervasive alternative splicing at the BRCA1 locus: A report from the ENIGMA consortium. Hum. Mol. Genet. 2014, 23, 3666–3680. [Google Scholar] [CrossRef] [PubMed]
- Gambino, G.; Tancredi, M.; Falaschi, E.; Aretini, P.; Caligo, M.A. Characterization of three alternative transcripts of the BRCA1 gene in patients with breast cancer and a family history of breast and/or ovarian cancer who tested negative for pathogenic mutations. Int. J. Mol. Med. 2015, 35, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Maglic, D.; Stovall, D.B.; Cline, J.M.; Fry, E.A.; Mallakin, A.; Taneja, P.; Caudell, D.L.; Willingham, M.C.; Sui, G.; Inoue, K. DMP1, a splice isoform of the tumour suppressor DMP1 locus, induces proliferation and progression of breast cancer. J. Pathol. 2015, 236, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Tschan, M.P.; Federzoni, E.A.; Haimovici, A.; Britschgi, C.; Moser, B.A.; Jin, J.; Reddy, V.A.; Sheeter, D.A.; Fischer, K.M.; Sun, P.; et al. Human DMTF1 antagonizes DMTF1 regulation of the p14(ARF) tumor suppressor and promotes cellular proliferation. Biochim. Biophys. Acta 2015, 1849, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Li, J.; Shi, J.; Sui, G. From General Aberrant Alternative Splicing in Cancers and Its Therapeutic Application to the Discovery of an Oncogenic DMTF1 Isoform. Int. J. Mol. Sci. 2017, 18, 191. [Google Scholar] [CrossRef] [PubMed]
- Fuller, S.J.; Sivarajah, K.; Sugden, P.H. ErbB receptors, their ligands, and the consequences of their activation and inhibition in the myocardium. J. Mol. Cell. Cardiol. 2008, 44, 831–854. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.; Ullrich, A.; McGuire, W. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Kwong, K.Y.; Hung, M.C. A novel splice variant of HER2 with increased transformation activity. Mol. Carcinog. 1998, 23, 62–68. [Google Scholar] [CrossRef]
- Marchini, C.; Gabrielli, F.; Iezzi, M.; Zenobi, S.; Montani, M.; Pietrella, L.; Kalogris, C.; Rossini, A.; Ciravolo, V.; Castagnoli, L.; et al. The human splice variant Delta16HER2 induces rapid tumor onset in a reporter transgenic mouse. PLoS ONE 2011, 6, e18727. [Google Scholar] [CrossRef] [PubMed]
- Alajati, A.; Sausgruber, N.; Aceto, N.; Duss, S.; Sarret, S.; Voshol, H.; Bonenfant, D.; Bentires-Alj, M. Mammary tumor formation and metastasis evoked by a HER2 splice variant. Cancer Res. 2013, 73, 5320–5327. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, F.; Tagliabue, E.; Campiglio, M.; Pupa, S.M.; Balsari, A.; Menard, S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr. Relat. Cancer 2006, 13, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, F.; Ghedini, G.C.; Koschorke, A.; Triulzi, T.; Dugo, M.; Gasparini, P.; Casalini, P.; Palladini, A.; Iezzi, M.; Lamolinara, A.; et al. Pathobiological implications of the d16HER2 splice variant for stemness and aggressiveness of HER2-positive breast cancer. Oncogene 2017, 36, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Druillennec, S.; Dorard, C.; Eychène, A. Alternative splicing in oncogenic kinases: From physiological functions to cancer. J. Nucleic Acids 2012, 2012, 639062. [Google Scholar] [CrossRef] [PubMed]
- Cittelly, D.M.; Das, P.M.; Salvo, V.A.; Fonseca, J.P.; Burow, M.E.; Jones, F.E. Oncogenic HER2Δ16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors. Carcinogenesis 2010, 31, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Huynh, F.C.; Jones, F.E. MicroRNA-7 inhibits multiple oncogenic pathways to suppress HER2Δ16 mediated breast tumorigenesis and reverse trastuzumab resistance. PLoS ONE 2014, 22, E114419. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A. Aberrant Splicing of Estrogen Receptor, HER2, and CD44 Genes in Breast Cancer. Genet. Epigenet. 2015, 7, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Lu, J.; Chen, H.; Werner, S.; Williams, L.T. The human fibroblast growth factor receptor genes: A common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol. Cell. Biol. 1991, 11, 4627–4634. [Google Scholar] [CrossRef] [PubMed]
- Madden, S.F.; Clarke, C.; Gaule, P.; Aherne, S.T.; O’Donovan, N.; Clynes, M.; Crown, J.; Gallagher, W.M. BreastMark: An integrated approach to mining publicly available transcriptomic datasets relating to breast cancer outcome. Breast Cancer Res. 2013, 15, R52. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, J.; Dutta, K.; Ilghari, D.; Beenken, A.; Goetz, R.; Eliseenkova, A.V.; Cowburn, D.; Mohammadi, M. The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. Structure 2012, 20, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Kan, M.; Yan, G.; Xu, J.; McKeehan, W.L. Alternately spliced NH2-terminal immunoglobulin-like loop I in the ectodomain of the fibroblast growth factor (FGF) receptor 1 lowers affinity for both heparin and FGF-1. J. Biol. Chem. 1995, 270, 10231–10235. [Google Scholar] [CrossRef] [PubMed]
- Luqmani, Y.A.; Mortimer, C.; Yiangou, C.; Johnston, C.L.; Bansal, G.S.; Sinnett, D.; Law, M.; Coombes, R.C. Expression of 2 variant forms of fibroblast growth factor receptor 1 in human breast. Int. J. Cancer 1995, 64, 274–279. [Google Scholar] [CrossRef] [PubMed]
- DiFeo, A.; Martignetti, J.A.; Narla, G. The role of KLF6 and its splice variants in cancer therapy. Drug Resist. Updates 2009, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vetter, D.; Cohen-Naftaly, M.; Villanueva, A.; Lee, Y.A.; Kocabayoglu, P.; Hannivoort, R.; Narla, G.; M Llovet, J.; Thung, S.N.; Friedman, S.L. Enhanced hepatocarcinogenesis in mouse models and human hepatocellular carcinoma by coordinate KLF6 depletion and increased messenger RNA splicing. Hepatology 2012, 56, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Hatami, R.; Sieuwerts, A.M.; Izadmehr, S.; Yao, Z.; Qiao, R.F.; Papa, L.; Look, M.P.; Smid, M.; Ohlssen, J.; et al. Levine AC KLF6-SV1 drives breast cancer metastasis and is associated with poor survival. Sci. Transl. Med. 2013, 5, 169ra12. [Google Scholar] [CrossRef] [PubMed]
- Olson, O.A.; Joyce, J.A. A Splicing Twist on Metastasis. Sci. Transl. Med. 2013, 5, 169fs2. [Google Scholar] [CrossRef] [PubMed]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.; O’Donovan, N.; Browne, B.; O’Shea, C.; Crown, J.; Hill, A.D.; McDermott, E.; O’Higgins, N.; Duffy, M.J. Expression of survivin and its splice variants survivin-2B and survivin-DeltaEx3 in breast cancer. Br. J. Cancer 2005, 92, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Toyoda, M.; Shinohara, H.; Okuda, J.; Watanabe, I.; Yamamoto, T.; Tanaka, K.; Tenjo, T.; Tanigawa, N. Expression of survivin correlates with apoptosis, proliferation, and angiogenesis during human colorectal tumorigenesis. Cancer 2001, 91, 2026–2032. [Google Scholar] [CrossRef]
- Pavlidou, A.; Kroupis, C.; Dimas, K. Association of survivin splice variants with prognosis and treatment of breast cancer. World J. Clin. Oncol. 2014, 5, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Bennit, H.F.; Turay, D. Early diagnostic value of survivin and its alternative splice variants in breast cancer. BMC Cancer 2014, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Blasberg, R. PET imaging of gene expression. Eur. J. Cancer 2002, 38, 2137–2146. [Google Scholar]
- Aylon, Y.; Oren, M. New plays in the p53 theater. Curr. Opin. Genet. Dev. 2011, 21, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell. Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A. Aberrant splicing of the DMP1-ARF-MDM2-p53 pathway in cancer. Int. J. Cancer 2016, 139, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Yoshida, H.; Kitagishi, Y.; Nishimura, Y.; Matsuda, S. Alternative splicings on p53, BRCA1 and PTEN genes involved in breast cancer. Biochem. Biophys. Res. Commun. 2011, 413, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [PubMed]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Harbeck, N.; Nap, M.; Molina, R.; Nicolini, A.; Senkus, E.; Cardoso, F. Clinical use of biomarkers in breast cancer: Updated guidelines from the European Group on Tumor Markers (EGTM). Eur. J. Cancer 2017, 75, 284–298. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P.; Klinck, R.; Bramard, A.; Inkel, L.; Dufresne-Martin, G.; Koh, C.; Gervais-Bird, J.; Lapointe, E.; Froehlich, U.; Durand, M.; et al. Identification of Alternative Splicing Markers for Breast Cancer. Cancer Res. 2008, 68, 9525. [Google Scholar] [CrossRef] [PubMed]
- Martensen, P.M.; Oka, K.; Christensen, L.; Rettenberger, P.M.; Petersen, H.H.; Christensen, A.; Chan, L.; Heegaard, C.W.; Andreasen, P.A. Breast carcinoma epithelial cells express a very low-density lipoprotein receptor variant lacking the O-linked glycosylation domain encoded by exon 16, but with full binding activity for serine proteinase/serpin complexes and Mr-40,000 receptor-associated protein. Eur. J. Biochem. 1997, 248, 583–591. [Google Scholar] [PubMed]
- Sciacca, L.; Mineo, R.; Pandini, G.; Murabito, A.; Vigneri, R.; Belfiore, A. In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene 2002, 21, 8240–8250. [Google Scholar] [CrossRef] [PubMed]
- Lapuk, A.; Marr, H.; Jakkula, L.; Pedro, H.; Bhattacharya, S.; Purdom, E.; Hu, Z.; Simpson, K.; Pachter, L.; Durinck, S.; et al. Exon-level microarray analyses identify alternative splicing programs in breast cancer. Mol. Cancer Res. 2010, 8, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Hoadley, K.A.; Parker, J.S.; Perou, C.M. Identification of mRNA isoform switching in breast cancer. BMC Genom. 2016, 17, 181. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.; Jearawiriyapaisarn, N.; Kole, R. Therapeutic potential of splice switching oligonucleotides. Oligonucleotides 2009, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zanetta, C.; Nizzardo, M.; Simone, C.; Monguzzi, E.; Bresolin, N.; Comi, G.P.; Corti, S. Molecular Therapeutic Strategies for Spinal Muscular Atrophies: Current and Future Clinical Trials. Clin. Ther. 2014, 36, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Zaharieva, E.; Chipman, J.K.; Soller, M. Alternative splicing interference by xenobiotics. Toxicology 2012, 296, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Salton, M.; Misteli, T. Small Molecule Modulators of Pre-mRNA Splicing in Cancer Therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.; Ranson, M.; Kelso, M.J. Anti-tumour/metastasis effects of the potassium-sparing diuretic amiloride: An orally active anti-cancer drug waiting for its call-of-duty? Int. J. Cancer 2011, 129, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.P.; Chao, C.C. Cancer cells acquire resistance to anticancer drugs: An update. Biomed. J. 2012, 35, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Froehlich, U.; Paquet, E.R.; Toutant, J.; Elela, S.A.; Chabot, B. Anticancer drugs affect the alternative splicing of Bcl-x and other human apoptotic genes. Mol. Cancer Ther. 2008, 7, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, M.; Delforge, Y.; Deward, A.; Habraken, Y.; Hennuy, B.; Piette, J.; Klinck, R.; Chabot, B.; Colige, A.; Lambert, C. Role of the splicing factor SRSF4 in cisplatin induced modifications of pre-mRNA splicing and apoptosis. BMC Cancer 2015, 15, 227. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.; Ruan, S.; Wang, M.; Habraken, Y.; Hennuy, B.; Piette, J.; Klinck, R.; Chabot, B.; Colige, A.; Lambert, C. A novel chemical, STF-083010, reverses tamoxifen-related drug resistance in breast cancer by inhibiting IRE1/XBP1. Oncotarget 2015, 6, 40692–40703. [Google Scholar] [CrossRef] [PubMed]
- Margolese, R.G.; Fisher, B.; Hortobagyi, G.N.; Buchholz, T.A. Neoplasms of the breast. In Cancer Medicine, 6th ed.; Section 32, Chapter 18; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Gansler, T.S., Holland, J.F., Frei, E., Eds.; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Stahlberg, C.; Pedersen, A.T.; Lynge, E.; Andersen, Z.J.; Keiding, N.; Hundrup, Y.A.; Obel, E.B.; Ottesen, B. Increased risk of breast cancer following different regimens of hormone replacement therapy frequently used in Europe. Int. J. Cancer 2004, 109, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Brown, M. Advances in estrogen receptor biology: Prospects for improvements in targeted breast cancer therapy. Breast Cancer Res. 2004, 6, 39–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chlebowski, R.T.; Anderson, G.L. Menopausal hormone therapy and cancer: Changing clinical observations of target site specificity. Steroids 2014, 90, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Begam, A.J.; Jubie, S.; Nanjan, M.J. Estrogen receptor agonists/antagonists in breast cancer therapy: A critical review. Bioorg. Chem. 2017, 71, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, L.H.; Williams, A.R.; Critchley, H.O. Selective progesterone receptor modulators. Curr. Opin. Obstet. Gynecol. 2014, 26, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Dorssers, L.C.; Van der Flier, S.; Brinkman, A.; van Agthoven, T.; Veldscholte, J.; Berns, E.M.; Klijn, J.G.; Beex, L.V.; Foekens, J.A. Tamoxifen resistance in breast cancer: Elucidating mechanisms. Drugs 2001, 61, 1721–1733. [Google Scholar] [CrossRef] [PubMed]
- Droog, M.; Beelen, K.; Linn, S.; Zwart, W. Tamoxifen resistance: From bench to bedside. Eur. J. Pharmacol. 2013, 717, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Aya, L.F.; Gonzalez-Angulo, A.M. Adjuvant systemic therapies in breast cancer. Surg. Clin. N. Am. 2013, 93, 473–491. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T. Clinical application of drug delivery systems in cancer chemotherapy: Review of the efficacy and side effects of approved drugs. Biol. Pharm. Bull. 2013, 36, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R.; Arpino, G.; Lee, A.S.; Hilsenbeck, V.G. Endocrine responsiveness: Understanding how progesterone receptor can be used to select endocrine therapy. Breast 2005, 14, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Iwase, H.; Yamamoto, Y. Clinical benefit of sequential use of endocrine therapies for metastatic breast cancer. Int. J. Clin. Oncol. 2015, 20, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Ribnikar, D.; Sousa, B.; Cufer, T.; Cardoso, F. Extended adjuvant endocrine therapy—A standard to all or some? Breast 2017, 32, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.C.; Cho, W.C.; Lin, P.W.; Lin, S.L.; Lee, W.Y.; Young, K.C. Quantitative profile of estrogen receptor variants/isoforms in Taiwanese women with breast cancer. Eur. J. Surg. Oncol. 2006, 32, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Ho, S.; Tarapore, P.; Chung, I.; Leung, Y.K. Estrogen Receptor β Isoform 5 Confers Sensitivity of Breast Cancer Cell Lines to Chemotherapeutic Agent-Induced Apoptosis through Interaction with Bcl2L12. Neoplasia 2013, 15, 1262–1271. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Montiel, N.; Anaya-Ruiz, M.; Pérez-Santos, M.; Martínez-Contreras, R.D. Alternative Splicing in Breast Cancer and the Potential Development of Therapeutic Tools. Genes 2017, 8, 217. https://doi.org/10.3390/genes8100217
Martínez-Montiel N, Anaya-Ruiz M, Pérez-Santos M, Martínez-Contreras RD. Alternative Splicing in Breast Cancer and the Potential Development of Therapeutic Tools. Genes. 2017; 8(10):217. https://doi.org/10.3390/genes8100217
Chicago/Turabian StyleMartínez-Montiel, Nancy, Maricruz Anaya-Ruiz, Martín Pérez-Santos, and Rebeca D. Martínez-Contreras. 2017. "Alternative Splicing in Breast Cancer and the Potential Development of Therapeutic Tools" Genes 8, no. 10: 217. https://doi.org/10.3390/genes8100217
APA StyleMartínez-Montiel, N., Anaya-Ruiz, M., Pérez-Santos, M., & Martínez-Contreras, R. D. (2017). Alternative Splicing in Breast Cancer and the Potential Development of Therapeutic Tools. Genes, 8(10), 217. https://doi.org/10.3390/genes8100217