Alternative Splicing of L-type CaV1.2 Calcium Channels: Implications in Cardiovascular Diseases

{kind=link}
{kind=link}
{kind=link}
Abstract
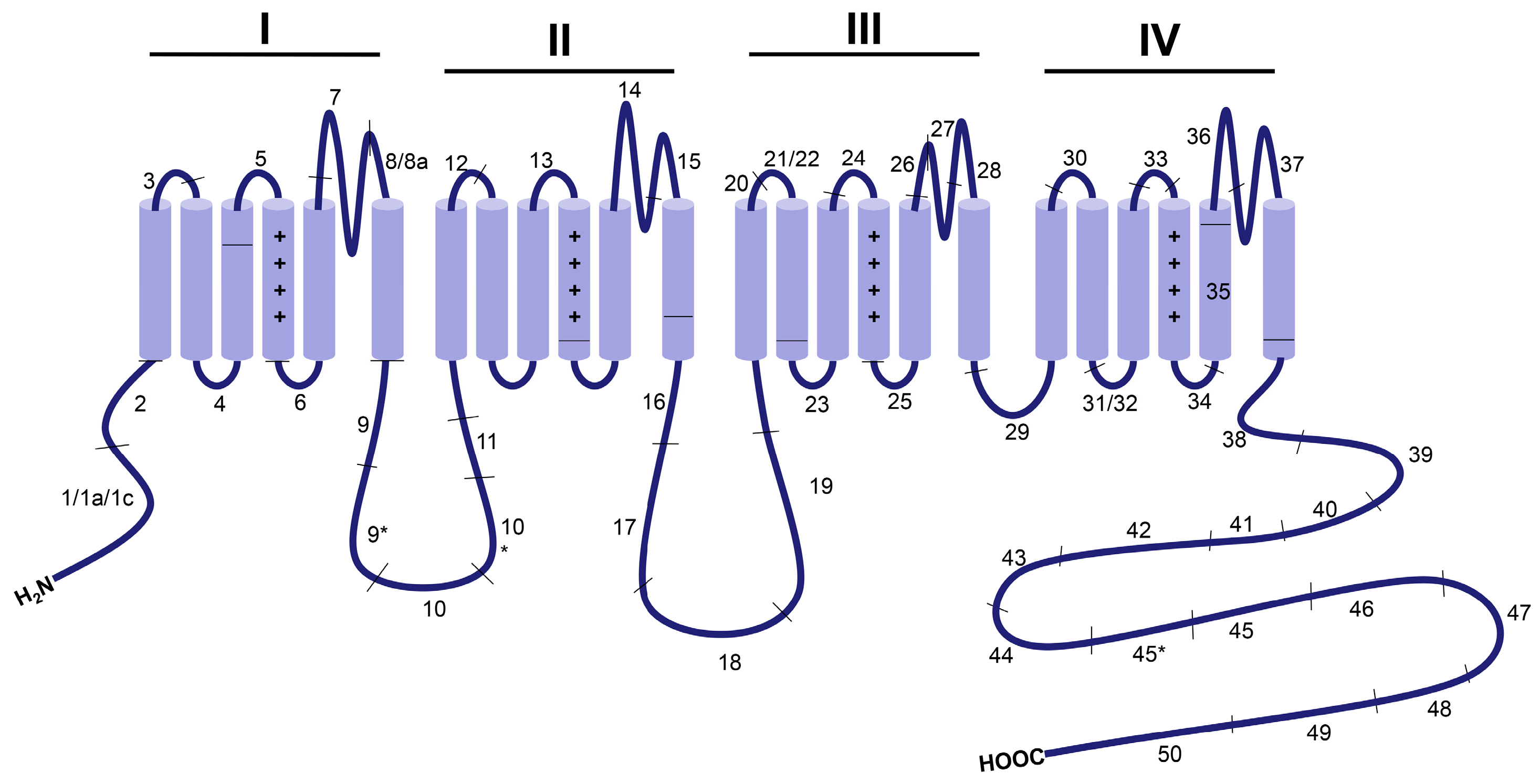
:1. Introduction
2. Overview of CaV1.2 Channel Function in the Cardiovascular System
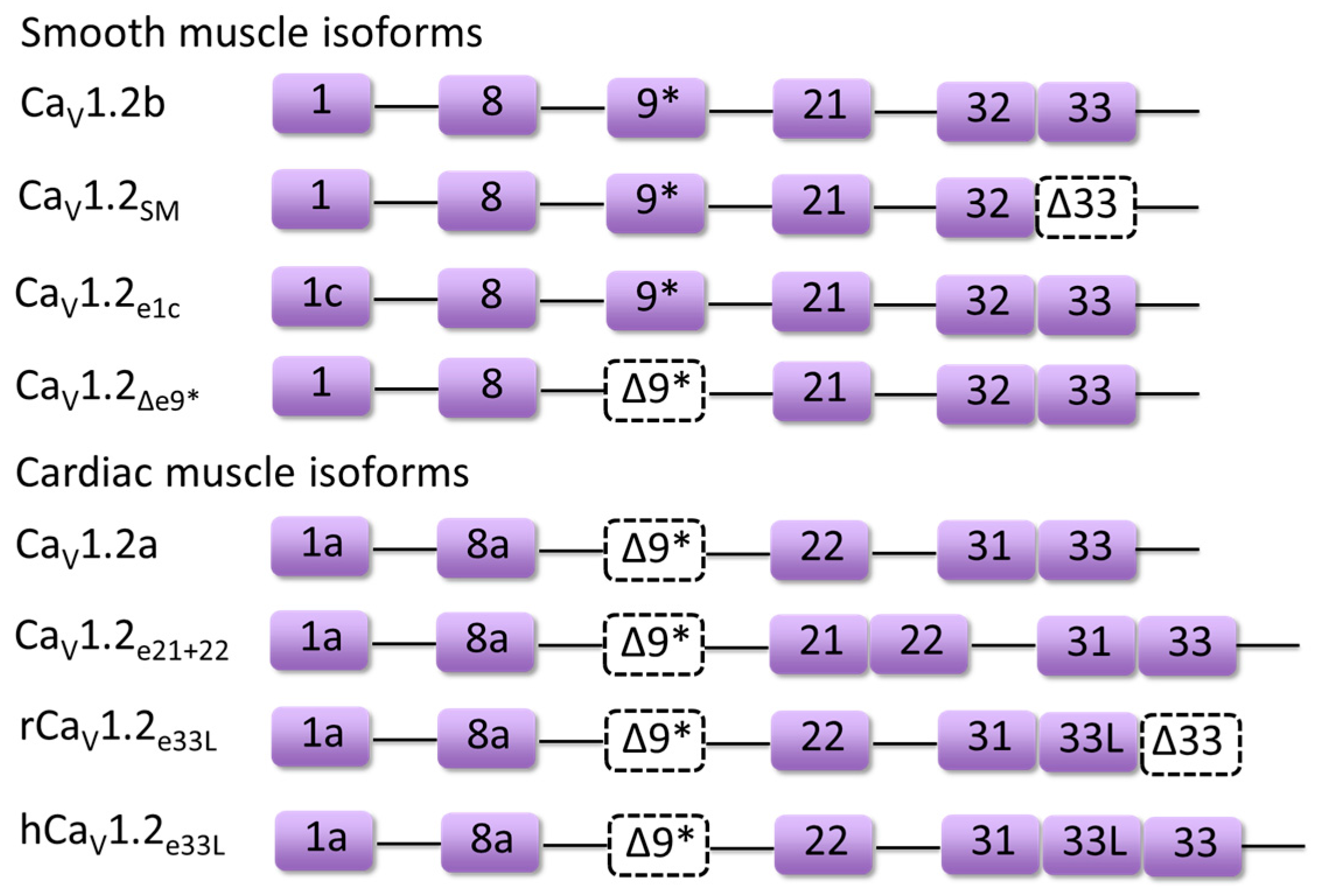
3. Developmentally Regulated Cardiac CaV1.2 Splicing
4. Cardiovascular Conditions-Related Splice Variants of CaV1.2 Channels
4.1. Timothy Syndrome
4.2. Heart Failure
4.3. Atherosclerosis
4.4. Hypertension
5. Splice Variant-Specific Modulation of CaV1.2 Channel Function
5.1. Modulation by CaV1.2-Interacting Partners
5.2. Modulation by Calcium Channel Blockers
6. Regulation of CaV1.2 Splicing by Splicing Factors
6.1. Rbfox Proteins
6.2. PTBP1
6.3. RBM20
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hofmann, F.; Flockerzi, V.; Kahl, S.; Wegener, J.W. L-type CaV1.2 calcium channels: From in vitro findings to in vivo function. Physiol. Rev. 2014, 94, 303–326. [Google Scholar] [CrossRef] [PubMed]
- Hell, J.W.; Westenbroek, R.E.; Warner, C.; Ahlijanian, M.K.; Prystay, W.; Gilbert, M.M.; Snutch, T.P.; Catterall, W.A. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell Biol. 1993, 123, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Welling, A.; Paparisto, S.; Hofmann, F.; Klugbauer, N. Enhanced expression of L-type Cav1.3 calcium channels in murine embryonic hearts from Cav1.2-deficient mice. J. Biol. Chem. 2003, 278, 40837–40841. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Yu, D.; Li, G.; Yong, T.F.; Soon, J.L.; Chua, Y.L.; Soong, T.W. A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J. Biol. Chem. 2007, 282, 35133–35142. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Goh, G.; Stolting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.N.; Berggren, P.O. The role of voltage-gated calcium channels in pancreatic β-cell physiology and pathophysiology. Endocr. Rev. 2006, 27, 621–676. [Google Scholar] [CrossRef] [PubMed]
- Striessnig, J.; Koschak, A.; Sinnegger-Brauns, M.J.; Hetzenauer, A.; Nguyen, N.K.; Busquet, P.; Pelster, G.; Singewald, N. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem. Soc. Trans. 2006, 34, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Brandt, A.; Striessnig, J.; Moser, T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J. Neurosci. 2003, 23, 10832–10840. [Google Scholar] [PubMed]
- Mangoni, M.E.; Couette, B.; Bourinet, E.; Platzer, J.; Reimer, D.; Striessnig, J.; Nargeot, J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. USA 2003, 100, 5543–5548. [Google Scholar] [CrossRef] [PubMed]
- Zampini, V.; Johnson, S.L.; Franz, C.; Lawrence, N.D.; Munkner, S.; Engel, J.; Knipper, M.; Magistretti, J.; Masetto, S.; Marcotti, W. Elementary properties of CaV1.3 Ca2+ channels expressed in mouse cochlear inner hair cells. J. Physiol. 2010, 588, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Baumann, L.; Gerstner, A.; Zong, X.; Biel, M.; Wahl-Schott, C. Functional characterization of the L-type Ca2+ channel Cav1.4alpha1 from mouse retina. Investig. Ophthalmol. Vis. Sci. 2004, 45, 708–713. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Bannister, J.P.; Adebiyi, A.; Zhao, G.; Narayanan, D.; Thomas, C.M.; Feng, J.Y.; Jaggar, J.H. Smooth muscle cell α2δ-1 subunits are essential for vasoregulation by CaV1.2 channels. Circ. Res. 2009, 105, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Bannister, J.P.; Bulley, S.; Narayanan, D.; Thomas-Gatewood, C.; Luzny, P.; Pachuau, J.; Jaggar, J.H. Transcriptional upregulation of α2δ-1 elevates arterial smooth muscle cell voltage-dependent Ca2+ channel surface expression and cerebrovascular constriction in genetic hypertension. Hypertension 2012, 60, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Altier, C.; Garcia-Caballero, A.; Simms, B.; You, H.; Chen, L.; Walcher, J.; Tedford, H.W.; Hermosilla, T.; Zamponi, G.W. The Cavβ subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat. Neurosci. 2011, 14, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Hill-Eubanks, D.C.; Werner, M.E.; Heppner, T.J.; Nelson, M.T. Calcium signaling in smooth muscle. Cold Spring Harb. Perspect. Biol. 2011, 3, a004549. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.M.; Colecraft, H.M. L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc. Res. 2013, 98, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Kharade, S.V.; Sonkusare, S.K.; Srivastava, A.K.; Thakali, K.M.; Fletcher, T.W.; Rhee, S.W.; Rusch, N.J. The β3 subunit contributes to vascular calcium channel upregulation and hypertension in angiotensin II-infused C57BL/6 mice. Hypertension 2013, 61, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Foell, J.D.; Balijepalli, R.C.; Delisle, B.P.; Yunker, A.M.; Robia, S.L.; Walker, J.W.; McEnery, M.W.; January, C.T.; Kamp, T.J. Molecular heterogeneity of calcium channel β-subunits in canine and human heart: Evidence for differential subcellular localization. Physiol. Genom. 2004, 17, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Nakayama, H.; Zhang, X.; Ai, X.; Harris, D.M.; Tang, M.; Zhang, H.; Szeto, C.; Stockbower, K.; Berretta, R.M.; et al. Calcium influx through Cav1.2 is a proximal signal for pathological cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2011, 50, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Z.; Liao, P.; Li, G.; Jiang, F.L.; Yu, D.; Hong, X.; Yong, T.F.; Tan, G.; Lu, S.; Wang, J.; et al. Differential splicing patterns of L-type calcium channel Cav1.2 subunit in hearts of Spontaneously Hypertensive Rats and Wistar Kyoto Rats. Biochim. Biophys. Acta 2008, 1783, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Yu, D.; Hu, Z.; Liang, M.C.; Wang, J.J.; Yu, C.Y.; Ng, G.; Yong, T.F.; Soon, J.L.; Chua, Y.L.; et al. Alternative splicing generates a novel truncated Cav1.2 channel in neonatal rat heart. J. Biol. Chem. 2015, 290, 9262–9272. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Zhang, H.Y.; Soong, T.W. Alternative splicing of voltage-gated calcium channels: From molecular biology to disease. Pflugers Arch. 2009, 458, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, X.; Margulies, K.; Jeevanandam, V.; Pollack, P.; Bailey, B.A.; Houser, S.R. L-type Ca2+ channel α1c subunit isoform switching in failing human ventricular myocardium. J. Mol. Cell. Cardiol. 2000, 32, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Yong, T.F.; Liang, M.C.; Yue, D.T.; Soong, T.W. Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovasc. Res. 2005, 68, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Schwoerer, A.P.; Neef, S.; Broichhausen, I.; Jacubeit, J.; Tiburcy, M.; Wagner, M.; Biermann, D.; Didie, M.; Vettel, C.; Maier, L.S.; et al. Enhanced Ca(2)+ influx through cardiac L-type Ca2+ channels maintains the systolic Ca2+ transient in early cardiac atrophy induced by mechanical unloading. Pflugers Arch. 2013, 465, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Best, J.M.; Kamp, T.J. Different subcellular populations of L-type Ca2+ channels exhibit unique regulation and functional roles in cardiomyocytes. J. Mol. Cell. Cardiol 2012, 52, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Goonasekera, S.A.; Hammer, K.; Auger-Messier, M.; Bodi, I.; Chen, X.; Zhang, H.; Reiken, S.; Elrod, J.W.; Correll, R.N.; York, A.J.; et al. Decreased cardiac L-type Ca2+ channel activity induces hypertrophy and heart failure in mice. J. Clin. Investig. 2012, 122, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Moosmang, S.; Schulla, V.; Welling, A.; Feil, R.; Feil, S.; Wegener, J.W.; Hofmann, F.; Klugbauer, N. Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 2003, 22, 6027–6034. [Google Scholar] [CrossRef] [PubMed]
- Pratt, P.F.; Bonnet, S.; Ludwig, L.M.; Bonnet, P.; Rusch, N.J. Upregulation of L-type Ca2+ channels in mesenteric and skeletal arteries of SHR. Hypertension 2002, 40, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Song, L.S.; Guia, A.; Muth, J.N.; Rubio, M.; Wang, S.Q.; Xiao, R.P.; Josephson, I.R.; Lakatta, E.G.; Schwartz, A.; Cheng, H. Ca2+ signaling in cardiac myocytes overexpressing the α1 subunit of L-type Ca2+ channel. Circ. Res. 2002, 90, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Makarewich, C.A.; Correll, R.N.; Gao, H.; Zhang, H.; Yang, B.; Berretta, R.M.; Rizzo, V.; Molkentin, J.D.; Houser, S.R. A caveolae-targeted L-type Ca2+ channel antagonist inhibits hypertrophic signaling without reducing cardiac contractility. Circ. Res. 2012, 110, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, A.V.; Balycheva, M.; Sanchez-Alonso, J.L.; Ilkan, Z.; Alvarez-Laviada, A.; Bhogal, N.; Diakonov, I.; Schobesberger, S.; Sikkel, M.B.; Bhargava, A.; et al. Direct Evidence for Microdomain-Specific Localization and Remodeling of Functional L-Type Calcium Channels in Rat and Human Atrial Myocytes. Circulation 2015, 132, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Correll, R.N.; Makarewich, C.A.; Zhang, H.; Zhang, C.; Sargent, M.A.; York, A.J.; Berretta, R.M.; Chen, X.; Houser, S.R.; Molkentin, J.D. Caveolae-localized L-type Ca2+ channels do not contribute to function or hypertrophic signalling in the mouse heart. Cardiovasc. Res. 2017, 113, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Tonegawa, K.; Otsuka, W.; Kumagai, S.; Matsunami, S.; Hayamizu, N.; Tanaka, S.; Moriwaki, K.; Obana, M.; Maeda, M.; Asahi, M.; et al. Caveolae-specific activation loop between CaMKII and L-type Ca2+ channel aggravates cardiac hypertrophy in α1-adrenergic stimulation. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H501–H514. [Google Scholar] [CrossRef] [PubMed]
- Diebold, R.J.; Koch, W.J.; Ellinor, P.T.; Wang, J.J.; Muthuchamy, M.; Wieczorek, D.F.; Schwartz, A. Mutually exclusive exon splicing of the cardiac calcium channel α 1 subunit gene generates developmentally regulated isoforms in the rat heart. Proc. Natl. Acad. Sci. USA 1992, 89, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Z.; Liang, M.C.; Lu, S.; Yu, D.; Yu, C.Y.; Yue, D.T.; Soong, T.W. Transcript scanning reveals novel and extensive splice variations in human l-type voltage-gated calcium channel, Cav1.2 alpha1 subunit. J. Biol. Chem. 2004, 279, 44335–44343. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, J.W.; Yu, D.; Soon, J.L.; de Kleijn, D.P.; Foo, R.; Liao, P.; Colecraft, H.M.; Soong, T.W. Aberrant Splicing Promotes Proteasomal Degradation of L-type CaV1.2 Calcium Channels by Competitive Binding for CaVbeta Subunits in Cardiac Hypertrophy. Sci. Rep. 2016, 6, 35247. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Decher, N.; Kumar, P.; Sachse, F.B.; Beggs, A.H.; Sanguinetti, M.C.; Keating, M.T. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 8089–8096. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Papp, A.C.; Binkley, P.F.; Johnson, J.A.; Sadee, W. Highly variable mRNA expression and splicing of L-type voltage-dependent calcium channel alpha subunit 1C in human heart tissues. Pharmacogenet. Genom. 2006, 16, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Dick, I.E.; Joshi-Mukherjee, R.; Yang, W.; Yue, D.T. Arrhythmogenesis in Timothy Syndrome is associated with defects in Ca2+-dependent inactivation. Nat. Commun. 2016, 7, 10370. [Google Scholar] [CrossRef] [PubMed]
- Wemhoner, K.; Friedrich, C.; Stallmeyer, B.; Coffey, A.J.; Grace, A.; Zumhagen, S.; Seebohm, G.; Ortiz-Bonnin, B.; Rinne, S.; Sachse, F.B.; et al. Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome. J. Mol. Cell. Cardiol. 2015, 80, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Li, G.; Yu, D.J.; Yong, T.F.; Wang, J.J.; Wang, J.; Soong, T.W. Molecular alteration of CaV1.2 calcium channel in chronic myocardial infarction. Pflugers Arch. 2009, 458, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, J.; Liao, P.; Bartels, P.; Zhang, H.; Yu, D.; Liang, M.C.; Poh, K.K.; Yu, C.Y.; Jiang, F.; et al. Exclusion of alternative exon 33 of CaV1.2 calcium channels in heart is proarrhythmogenic. Proc. Natl. Acad. Sci. USA 2017, 114, E4288–E4295. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Zhang, Y.; Heller, J.; Abernethy, D.R.; Soldatov, N.M. Atherosclerosis-related molecular alteration of the human CaV1.2 calcium channel α1C subunit. Proc. Natl. Acad. Sci. USA 2006, 103, 17024–17029. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; et al. Executive Summary: Heart Disease and Stroke Statistics--2016 Update: A Report From the American Heart Association. Circulation 2016, 133, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Bannister, J.P.; Thomas-Gatewood, C.M.; Neeb, Z.P.; Adebiyi, A.; Cheng, X.; Jaggar, J.H. Ca(V)1.2 channel N-terminal splice variants modulate functional surface expression in resistance size artery smooth muscle cells. J. Biol. Chem. 2011, 286, 15058–15066. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fan, J.; Zhu, H.; Ji, L.; Fan, W.; Kapoor, I.; Wang, Y.; Zhu, G.; Wang, J. Aberrant Splicing Induced by Dysregulated Rbfox2 Produces Enhanced Function of CaV1.2 Calcium Channel and Vascular Myogenic Tone in Hypertension. Hypertension 2017, 70, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Nystoriak, M.A.; Murakami, K.; Penar, P.L.; Wellman, G.C. CaV1.2 splice variant with exon 9* is critical for regulation of cerebral artery diameter. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1820–H1828. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Yu, D.; Lu, S.; Tang, Z.; Liang, M.C.; Zeng, S.; Lin, W.; Soong, T.W. Smooth muscle-selective alternatively spliced exon generates functional variation in Cav1.2 calcium channels. J. Biol. Chem. 2004, 279, 50329–50335. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.H.; Fromme, S. Expression of Calcium Channel Subunit Variants in Small Mesenteric Arteries of WKY and SHR. Am. J. Hypertens. 2015, 28, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Z.; Saada, N.; Dai, B.; Pang, L.; Palade, P. Vascular-specific increase in exon 1B-encoded CAV1.2 channels in spontaneously hypertensive rats. Am. J. Hypertens. 2006, 19, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Camby, I.; Le Mercier, M.; Lefranc, F.; Kiss, R. Galectin-1: A small protein with major functions. Glycobiology 2006, 16, 137R–157R. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Thio, S.S.; Yang, S.S.; Yu, D.; Yu, C.Y.; Wong, Y.P.; Liao, P.; Li, S.; Soong, T.W. Splice Variant Specific Modulation of Cav1.2 Calcium Channel by Galectin-1 Regulates Arterial Constriction. Circ. Res. 2011, 109, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C. The α2δ subunits of voltage-gated calcium channels. Biochim. Biophys. Acta 2013, 1828, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Liao, P.; Wang, J.J.; Yu, D.J.; Soong, T.W. Alternative splicing modulates diltiazem sensitivity of cardiac and vascular smooth muscle CaV1.2 calcium channels. Br. J. Pharmacol. 2010, 160, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Z.; Zheng, S.; Nikolic, J.; Black, D.L. Developmental control of CaV1.2 L-type calcium channel splicing by Fox proteins. Mol. Cell. Biol. 2009, 29, 4757–4765. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, G.; Yu, D.; Wong, Y.P.; Yong, T.F.; Liang, M.C.; Liao, P.; Foo, R.; Hoppe, U.C.; Soong, T.W. Characterization of CaV1.2 exon 33 heterozygous knockout mice and negative correlation between Rbfox1 and CaV1.2 exon 33 expressions in human heart failure. Channels 2017. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Ren, S.; Lee, J.H.; Qiu, J.; Chapski, D.J.; Rau, C.D.; Zhou, Y.; Abdellatif, M.; Nakano, A.; Vondriska, T.M.; et al. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J. Clin. Investig. 2016, 126, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Z.; Sharma, S.; Zheng, S.; Chawla, G.; Nikolic, J.; Black, D.L. Regulation of the mutually exclusive exons 8a and 8 in the CaV1.2 calcium channel transcript by polypyrimidine tract-binding protein. J. Biol. Chem. 2011, 286, 10007–10016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bahi, N.; Llovera, M.; Comella, J.X.; Sanchis, D. Polypyrimidine tract binding proteins (PTB) regulate the expression of apoptotic genes and susceptibility to caspase-dependent apoptosis in differentiating cardiomyocytes. Cell Death Differ. 2009, 16, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, W.; Dewey, C.N.; Greaser, M.L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013, 41, 2659–2672. [Google Scholar] [CrossRef] [PubMed]
- Refaat, M.M.; Lubitz, S.A.; Makino, S.; Islam, Z.; Frangiskakis, J.M.; Mehdi, H.; Gutmann, R.; Zhang, M.L.; Bloom, H.L.; MacRae, C.A.; et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm 2012, 9, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Liang, M.C.; Soong, T.W. Alternative Splicing of L-type CaV1.2 Calcium Channels: Implications in Cardiovascular Diseases. Genes 2017, 8, 344. https://doi.org/10.3390/genes8120344
Hu Z, Liang MC, Soong TW. Alternative Splicing of L-type CaV1.2 Calcium Channels: Implications in Cardiovascular Diseases. Genes. 2017; 8(12):344. https://doi.org/10.3390/genes8120344
Chicago/Turabian StyleHu, Zhenyu, Mui Cheng Liang, and Tuck Wah Soong. 2017. "Alternative Splicing of L-type CaV1.2 Calcium Channels: Implications in Cardiovascular Diseases" Genes 8, no. 12: 344. https://doi.org/10.3390/genes8120344
APA StyleHu, Z., Liang, M. C., & Soong, T. W. (2017). Alternative Splicing of L-type CaV1.2 Calcium Channels: Implications in Cardiovascular Diseases. Genes, 8(12), 344. https://doi.org/10.3390/genes8120344