Epigenetic Basis of Cellular Senescence and Its Implications in Aging

Abstract
:1. Cellular Senescence: A Double-Edged Sword against Cancer
2. Epigenetic Changes during Cellular Senescence and Aging
2.1. Heterochromatin Changes Accompany Cellular Senescence
2.2. Senescence-Associated Distention of Satellites Is a Senescence-Associated Heterochromatic Foci -Independent Epigenetic Change
3. Epigenetic Effectors of Cellular Senescence
3.1. The Senescence-Associated Heterochromatic Foci and High Mobility Group Proteins Cooperate for the Senescence Phenotype
3.2. Epigenetic Regulators of the Senescence Associated Secretory Phenotype
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16(INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving atm, p53, and p21CIP1, but not p16INK4a. Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Itahana, K.; Dimri, G.; Campisi, J. Regulation of cellular senescence by p53. Eur. J. Biochem. 2001, 268, 2784–2791. [Google Scholar] [CrossRef] [PubMed]
- Tonnessen-Murray, C.A.; Lozano, G.; Jackson, J.G. The regulation of cellular functions by the p53 protein: Cellular senescence. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Janzen, V.; Forkert, R.; Fleming, H.E.; Saito, Y.; Waring, M.T.; Dombkowski, D.M.; Cheng, T.; DePinho, R.A.; Sharpless, N.E.; Scadden, D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 2006, 443, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Ramsey, M.R.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N.E. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.E.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. Notch1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.R.; Coppe, J.P.; Campisi, J.; Desprez, P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Fougere, B.; Boulanger, E.; Nourhashemi, F.; Guyonnet, S.; Cesari, M. Chronic inflammation: Accelerator of biological aging. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef] [PubMed]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.; Putavet, D.A.; Klein, J.D.; Derks, K.W.; Bourgeois, B.R.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Campisi, J.; Dimri, G.P. Methods to detect biomarkers of cellular senescence: The senescence-associated β-galactosidase assay. Methods Mol. Biol. 2007, 371, 21–31. [Google Scholar] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Kovatcheva, M.; Liao, W.; Klein, M.E.; Robine, N.; Geiger, H.; Crago, A.M.; Dickson, M.A.; Tap, W.D.; Singer, S.; Koff, A. ATRX is a regulator of therapy induced senescence in human cells. Nat. Commun. 2017, 8, 386. [Google Scholar] [CrossRef] [PubMed]
- Kreiling, J.A.; Tamamori-Adachi, M.; Sexton, A.N.; Jeyapalan, J.C.; Munoz-Najar, U.; Peterson, A.L.; Manivannan, J.; Rogers, E.S.; Pchelintsev, N.A.; Adams, P.D.; et al. Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell 2011, 10, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.R.; Xu, M.; Kinzler, K.A.; Kaur, A.; Appleton, J.; O’Connell, M.P.; Marchbank, K.; Valiga, A.; Dang, V.M.; Perego, M.; et al. Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell Melanoma Res. 2015, 28, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini Denchi, E.; Attwooll, C.; Pasini, D.; Helin, K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol. Cell. Biol. 2005, 25, 2660–2672. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Sadaie, M.; Salama, R.; Carroll, T.; Tomimatsu, K.; Chandra, T.; Young, A.R.; Narita, M.; Perez-Mancera, P.A.; Bennett, D.C.; Chong, H.; et al. Redistribution of the lamin b1 genomic binding profile affects rearrangement of heterochromatic domains and sahf formation during senescence. Genes Dev. 2013, 27, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Kosar, M.; Bartkova, J.; Hubackova, S.; Hodny, Z.; Lukas, J.; Bartek, J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16ink4a. Cell Cycle 2011, 10, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.Y.; Andrews, S.; Fraser, P.; Reik, W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015, 10, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Criscione, S.W.; De Cecco, M.; Siranosian, B.; Zhang, Y.; Kreiling, J.A.; Sedivy, J.M.; Neretti, N. Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2016, 2, e1500882. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Kirschner, K. Chromosome organisation during ageing and senescence. Curr. Opin. Cell Biol. 2016, 40, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, A.; Li, W.X. Global heterochromatin loss: A unifying theory of aging? Epigenetics 2012, 7, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chang, W.Y.; Etheridge, A.; Strickfaden, H.; Jin, Z.; Palidwor, G.; Cho, J.H.; Wang, K.; Kwon, S.Y.; Dore, C.; et al. Reprogramming progeria fibroblasts re-establishes a normal epigenetic landscape. Aging Cell 2017, 16, 870–887. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.A.; Neretti, N.; Sedivy, J.M. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 2013, 12, 247–256. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Criscione, S.W.; Peterson, A.L.; Neretti, N.; Sedivy, J.M.; Kreiling, J.A. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging 2013, 5, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Helfand, S.L. Chromatin structure and transposable elements in organismal aging. Front. Genet. 2013, 4, 274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chen, W.; Adams, P.D. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell. Biol. 2007, 27, 2343–2358. [Google Scholar] [CrossRef] [PubMed]
- Swanson, E.C.; Manning, B.; Zhang, H.; Lawrence, J.B. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol. 2013, 203, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Fraser, D.G.; Wang, H.; Jaehn, K.; Ogrodnik, M.B.; Weivoda, M.M.; Drake, M.T.; Tchkonia, T.; LeBrasseur, N.K.; Kirkland, J.L.; et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res. 2016, 31, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Dillinger, S.; Straub, T.; Nemeth, A. Nucleolus association of chromosomal domains is largely maintained in cellular senescence despite massive nuclear reorganisation. PLoS ONE 2017, 12, e0178821. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; Vanderkraats, N.D.; Shah, P.P.; van Tuyn, J.; Singh Rai, T.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Enukashvily, N.I.; Donev, R.; Waisertreiger, I.S.; Podgornaya, O.I. Human chromosome 1 satellite 3 DNA is decondensed, demethylated and transcribed in senescent cells and in A431 epithelial carcinoma cells. Cytogenet. Genome Res. 2007, 118, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Tasselli, L.; Xi, Y.; Zheng, W.; Tennen, R.I.; Odrowaz, Z.; Simeoni, F.; Li, W.; Chua, K.F. SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat. Struct. Mol. Biol. 2016, 23, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Hock, R.; Furusawa, T.; Ueda, T.; Bustin, M. HMG chromosomal proteins in development and disease. Trends Cell Biol. 2007, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Funayama, R.; Saito, M.; Tanobe, H.; Ishikawa, F. Loss of linker histone H1 in cellular senescence. J. Cell Biol. 2006, 175, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nunez, S.; Chicas, A.; Hearn, S.A.; Myers, M.P.; Lowe, S.W. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006, 126, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.P.; Rodier, F.; Campisi, J. p53-dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Tanizawa, H.; Fatkhutdinov, N.; Bitler, B.G.; Le, L.; Alicea, G.; Yang, T.L.; Johnson, F.B.; et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J. Cell Biol. 2016, 215, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, N.; Banito, A.; Roe, J.S.; Alonso-Curbelo, D.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.H.; Aksoy, O.; Bolden, J.E.; Chen, C.C.; et al. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 2016, 6, 612–629. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and atm play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef] [PubMed]
- Capell, B.C.; Drake, A.M.; Zhu, J.; Shah, P.P.; Dou, Z.; Dorsey, J.; Simola, D.F.; Donahue, G.; Sammons, M.; Rai, T.S.; et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 2016, 30, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Imai, Y.; Yamakoshi, K.; Kuninaka, S.; Ohtani, N.; Yoshimoto, S.; Hori, S.; Tachibana, M.; Anderton, E.; Takeuchi, T.; et al. DNA damage signaling triggers degradation of histone methyltransferases through APC/CCdh1 in senescent cells. Mol. Cell 2012, 45, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Nam, Y.; Koo, J.Y.; Lim, D.; Park, J.; Ock, J.; Kim, J.; Suk, K.; Park, S.B. A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nat. Chem. Biol. 2014, 10, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhang, W.; Yang, G.; Li, H.; Chen, Q.; Song, R.; Zhao, L. HMGB1 overexpression as a prognostic factor for survival in cancer: A meta-analysis and systematic review. Oncotarget 2016, 7, 50417–50427. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.H.; Kim, J.; Park, J.Y.; Hong, S.M.; Park, C.W.; Hong, S.J.; Park, S.Y.; Choi, Y.J.; Do, I.G.; Joh, J.W.; et al. Overexpression of high-mobility group box 2 is associated with tumor aggressiveness and prognosis of hepatocellular carcinoma. Clin. Cancer Res. 2010, 16, 5511–5521. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Hoshii, T.; Armstrong, S.A. Mixed-lineage leukemia fusions and chromatin in leukemia. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.C.; Dou, Y. Hijacked in cancer: The MLL/KMT2 family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Gelato, K.A.; Fernandez-Montalvan, A.; Siegel, S.; Haendler, B. Targeting BET bromodomains for cancer treatment. Epigenomics 2015, 7, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Gan, W.; Li, X.; Wang, S.; Zhang, W.; Huang, L.; Liu, S.; Zhong, Q.; Guo, J.; Zhang, J.; et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 2017, 23, 1063–1071. [Google Scholar] [CrossRef] [PubMed]


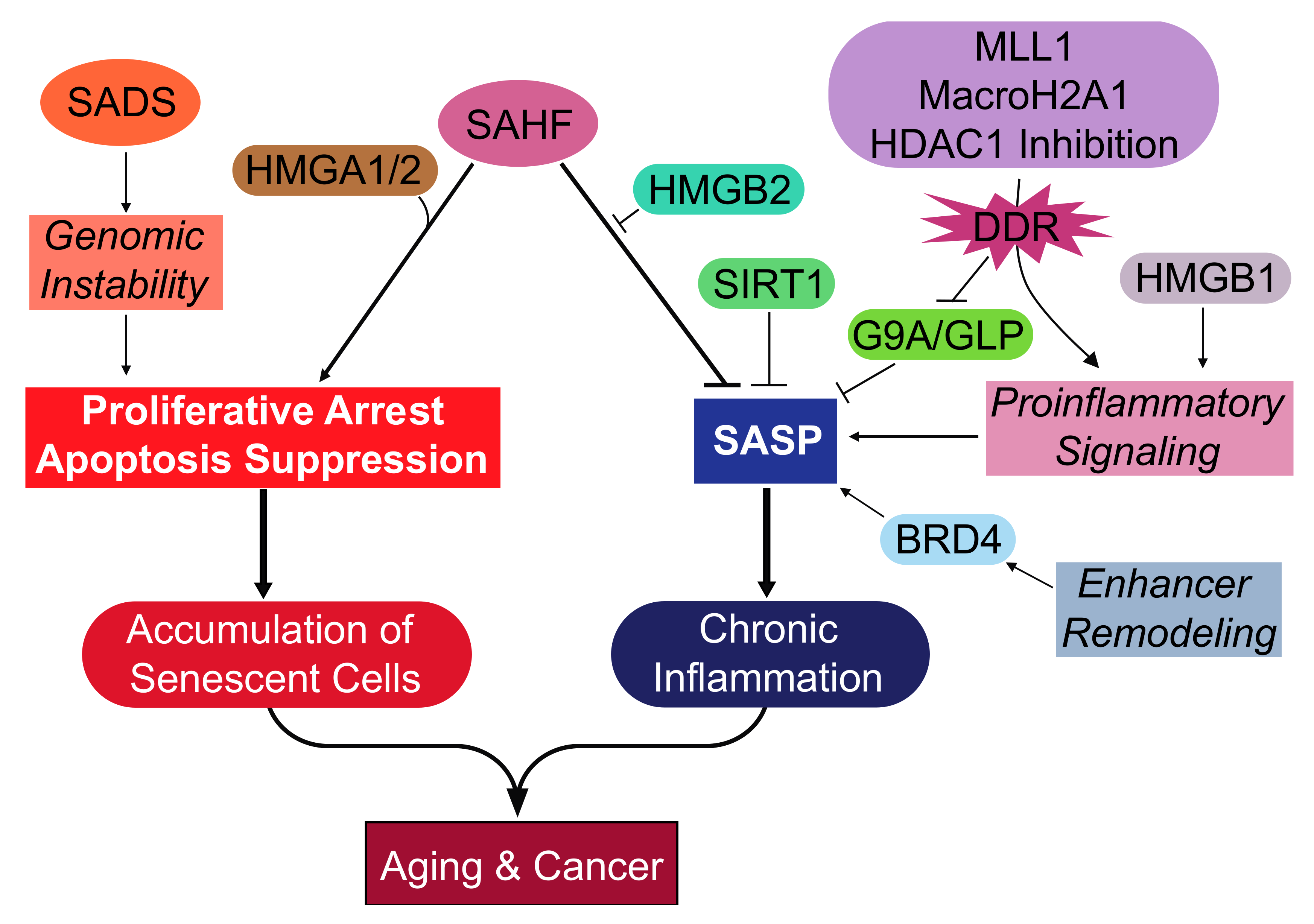{kind=link}
| Heterochromatin Modification or Activated Effector | Mode of Cellular Senescence | Function during Cellular Senescence | Cell Type | Reference |
|---|---|---|---|---|
| SAHF | Replicative Oncogene | Proliferative Arrest | Human Fibroblasts | [36,37,61] |
| SADS | Replicative Oncogene HGPS | Not Defined | Human Fibroblasts | [58,62] |
| Loss of Lamin B1 | Replicative Oncogene HGPS Genotoxic Stress: Irradiation | Proliferative Arrest | Human Fibroblasts | [51,52,53] |
| HMGA1/2 | Replicative Oncogene | Proliferative Arrest | Human Fibroblasts | [70] |
| HMGB1 | Replicative Oncogene Genotoxic stress: Irradiation | SASP Activation | Human Fibroblasts | [72] |
| HMGB2 | Oncogene | SASP Activation | Human Fibroblasts | [73] |
| HDAC1 | Genotoxic stress: Bleomycin HDAC Inhibitors: Sodium butyrate and Trichostatin A | SASP Activation | Human Fibroblasts | [79] |
| Enhancer Remodeling: BRD4 | Oncogene Genotoxic stress: Etoposide | SASP Activation | Human Fibroblasts | [74] |
| MLL1 | Oncogene Genotoxic stress: Etoposide | SASP Activation | Human Fibroblasts MCF7 human breast cancer cell line | [77] |
| MacroH2A1 | Oncogene | SASP Activation | Human Fibroblasts | [75] |
| G9a/GLP | Replicative Oncogene | SASP Inhibition | Human Fibroblasts | [78] |
| SIRT1 | Genotoxic stress: Irradiation | SASP Inhibition | Human Fibroblasts | [76] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nacarelli, T.; Liu, P.; Zhang, R. Epigenetic Basis of Cellular Senescence and Its Implications in Aging. Genes 2017, 8, 343. https://doi.org/10.3390/genes8120343
Nacarelli T, Liu P, Zhang R. Epigenetic Basis of Cellular Senescence and Its Implications in Aging. Genes. 2017; 8(12):343. https://doi.org/10.3390/genes8120343
Chicago/Turabian StyleNacarelli, Timothy, Pingyu Liu, and Rugang Zhang. 2017. "Epigenetic Basis of Cellular Senescence and Its Implications in Aging" Genes 8, no. 12: 343. https://doi.org/10.3390/genes8120343
APA StyleNacarelli, T., Liu, P., & Zhang, R. (2017). Epigenetic Basis of Cellular Senescence and Its Implications in Aging. Genes, 8(12), 343. https://doi.org/10.3390/genes8120343