
Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration


Abstract
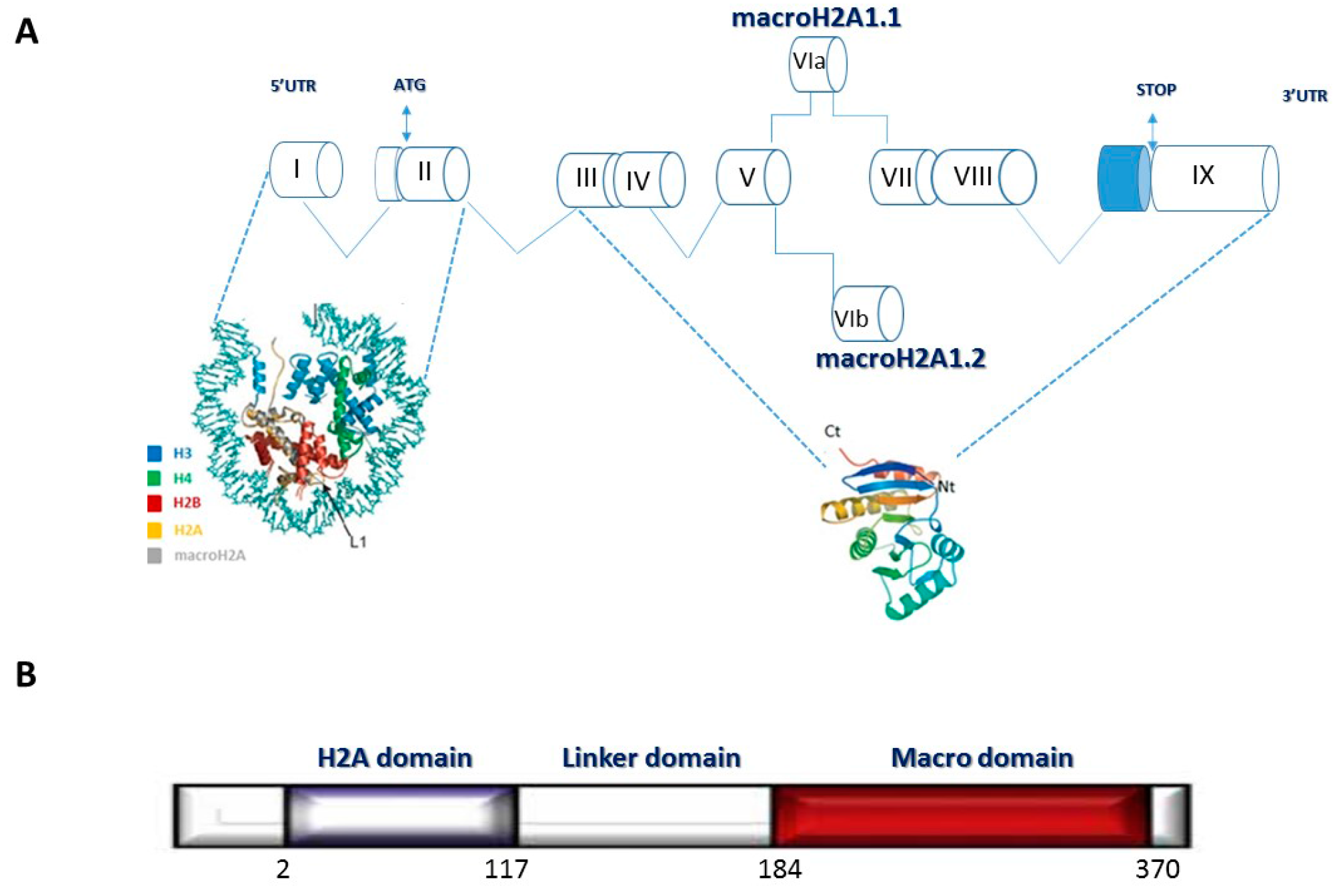
:1. Introduction
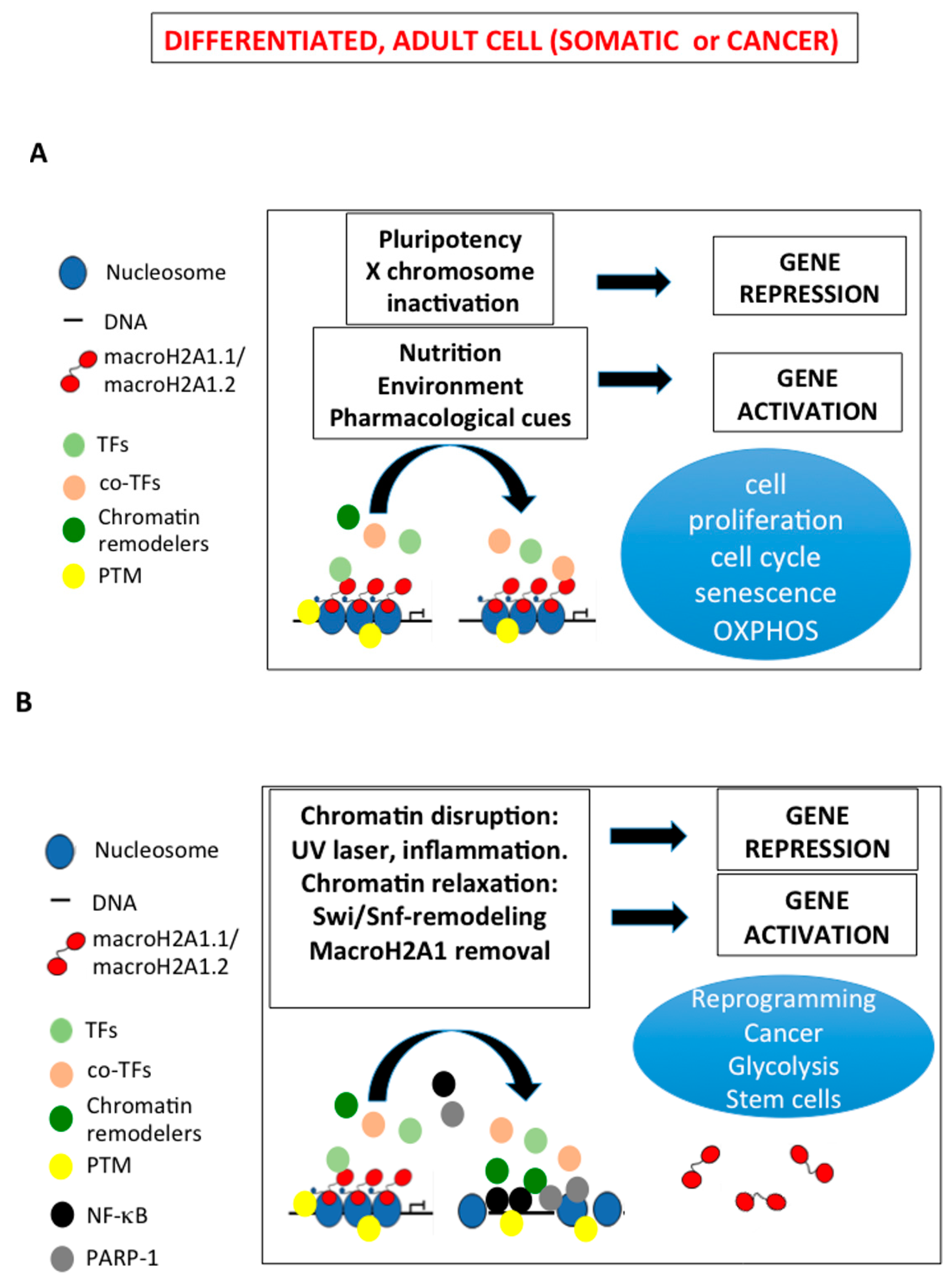
2. MacroH2A1, Cancer and Cellular Senescence
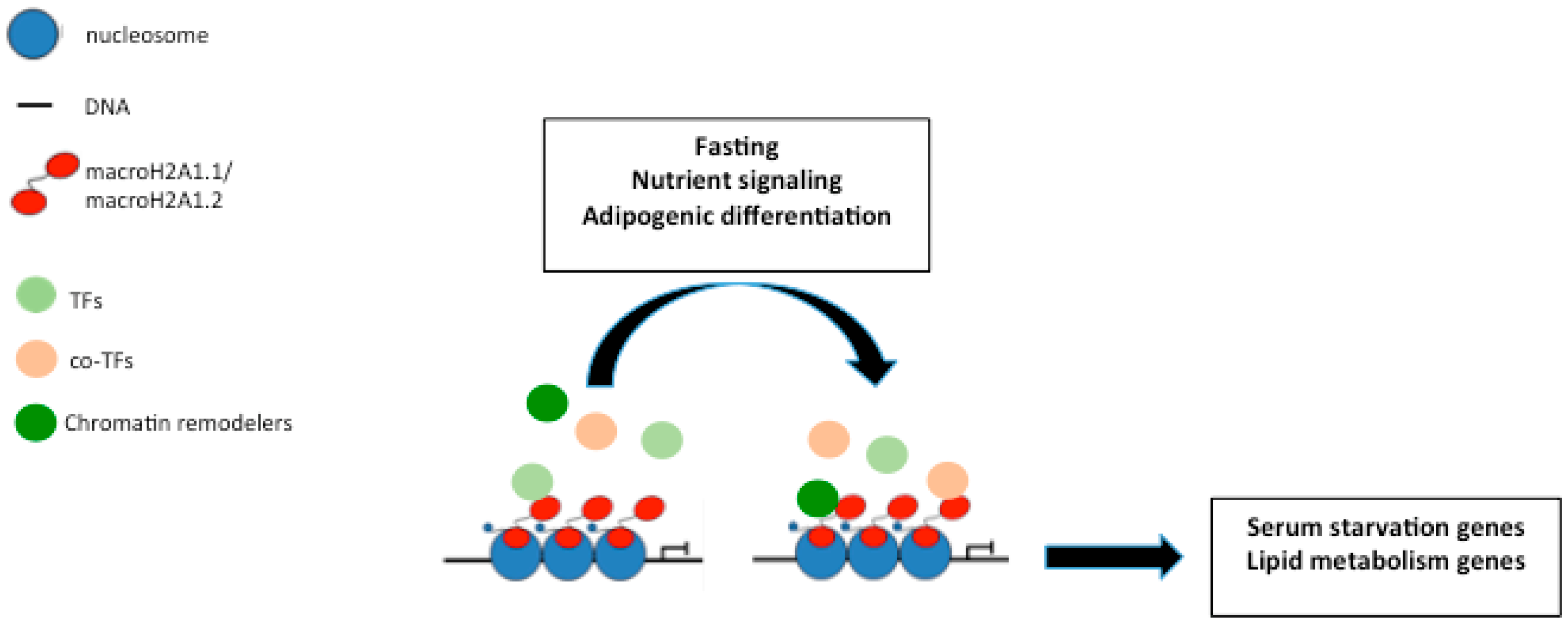
3. MacroH2A1 and Nutrients
4. MacroH2A1 and Cell Reprogramming
5. MacroH2A1 and Integration of Nutritional Signaling into Cell Proliferation and Fate
6. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Histone variants—Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Environmental responses mediated by histone variants. Trends Cell Biol. 2014, 24, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, S. Histone variants in pluripotency and disease. Development 2013, 140, 2513–2524. [Google Scholar] [CrossRef] [PubMed]
- Filipescu, D.; Muller, S.; Almouzni, G. Histone H3 variants and their chaperones during development and disease: Contributing to epigenetic control. Annu. Rev. Cell Dev. Biol. 2014, 30, 615–646. [Google Scholar] [CrossRef] [PubMed]
- Gurard-Levin, Z.A.; Quivy, J.P.; Almouzni, G. Histone chaperones: Assisting histone traffic and nucleosome dynamics. Annu. Rev. Biochem. 2014, 83, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Casas, C.; Gonzalez-Romero, R.; Cheema, M.S.; Ausio, J.; Eirin-Lopez, J.M. The characterization of macroH2A beyond vertebrates supports an ancestral origin and conserved role for histone variants in chromatin. Epigenetics 2016, 11, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.P.; Huang, T.; Mastrangelo, M.A.; Loring, J.; Panning, B.; Jaenisch, R. Messenger RNAs encoding mouse histone macroH2A1 isoforms are expressed at similar levels in male and female cells and result from alternative splicing. Nucleic Acids Res. 1999, 27, 3685–3689. [Google Scholar] [CrossRef] [PubMed]
- Kustatscher, G.; Hothorn, M.; Pugieux, C.; Scheffzek, K.; Ladurner, A.G. Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol. 2005, 12, 624–625. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Munoz, I.; Lund, A.H.; van der Stoop, P.; Boutsma, E.; Muijrers, I.; Verhoeven, E.; Nusinow, D.A.; Panning, B.; Marahrens, Y.; van Lohuizen, M. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 7635–7640. [Google Scholar] [CrossRef] [PubMed]
- Nusinow, D.A.; Hernandez-Munoz, I.; Fazzio, T.G.; Shah, G.M.; Kraus, W.L.; Panning, B. Poly(ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, MacroH2A, and contributes to silencing of the inactive X chromosome. J. Biol. Chem. 2007, 282, 12851–12859. [Google Scholar] [CrossRef] [PubMed]
- Mietton, F.; Sengupta, A.K.; Molla, A.; Picchi, G.; Barral, S.; Heliot, L.; Grange, T.; Wutz, A.; Dimitrov, S. Weak but uniform enrichment of the histone variant macroH2A1 along the inactive X chromosome. Mol. Cell Biol. 2009, 29, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Soma, A.; Sato, K.; Nakanishi, T. Visualization of inactive X chromosome in preimplantation embryos utilizing MacroH2A-EGFP transgenic mouse. Genesis 2013, 51, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Borghesan, M.; Fusilli, C.; Rappa, F.; Panebianco, C.; Rizzo, G.; Oben, J.A.; Mazzoccoli, G.; Faulkes, C.; Pata, I.; Agodi, A. DNA Hypomethylation and histone variant macroH2A1 synergistically attenuate chemotherapy-induced senescence to promote hepatocellular carcinoma progression. Cancer Res. 2016, 76, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Pazienza, V.; Panebianco, C.; Rappa, F.; Memoli, D.; Borghesan, M.; Cannito, S.; Oji, A.; Mazza, G.; Tamburrino, D.; Fusai, G. Histone macroH2A1.2 promotes metabolic health and leanness by inhibiting adipogenesis. Epigenet. Chromat. 2016, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Creppe, C.; Janich, P.; Cantarino, N.; Noguera, M.; Valero, V.; Musulen, E.; Douet, J.; Posavec, M.; Martín-Caballero, J.; Sumoy, L.; et al. MacroH2A1 regulates the balance between self-renewal and differentiation commitment in embryonic and adult stem cells. Mol. Cell Biol. 2012, 32, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Creppe, C.; Posavec, M.; Douet, J.; Buschbeck, M. MacroH2A in stem cells: A story beyond gene repression. Epigenomics 2012, 4, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Gamble, M.J.; Frizzell, K.M.; Yang, C.; Krishnakumar, R.; Kraus, W.L. The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. 2010, 24, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Cantarino, N.; Douet, J.; Buschbeck, M. MacroH2A—An epigenetic regulator of cancer. Cancer Lett. 2013, 336, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Posavec, M.; Timinszky, G.; Buschbeck, M. Macro domains as metabolite sensors on chromatin. Cell Mol. Life Sci. 2013, 70, 1509–1524. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, O.; Fusilli, C.; Rappa, F.; Van Haele, M.; Douet, J.; Pindjakova, J.; Rocha, S.W.; Pata, I.; Valčíková, B.; Uldrijan, S.; et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology 2017. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Pierredon, S.; Driouch, K.; Gratadou, L.; Lacroix-Triki, M.; Espinoza, M.P.; Zonta, E.; Germann, S.; Mortada, H.; Villemin, J.P.; et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 2012, 19, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Shim, J.W.; Park, H.S.; Eum, D.Y.; Park, M.T.; Mi Yi, J.; Choi, S.H.; Kim, S.D.; Son, T.G.; Lu, W.; et al. MacroH2A1 downregulation enhances the stem-like properties of bladder cancer cells by transactivation of Lin28B. Oncogene 2016, 35, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Novikov, L.; Park, J.W.; Chen, H.; Klerman, H.; Jalloh, A.S.; Gamble, M.J. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol. Cell Biol. 2011, 31, 4244–4255. [Google Scholar] [CrossRef] [PubMed]
- Sporn, J.C.; Kustatscher, G.; Hothorn, T.; Collado, M.; Serrano, M.; Muley, T.; Schnabel, P.; Ladurner, A.G. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 2009, 28, 3423–3428. [Google Scholar] [CrossRef] [PubMed]
- Jueliger, S.; Lyons, J.; Cannito, S.; Pata, I.; Pata, P.; Shkolnaya, M.; Lo Re, O.; Peyrou, M.; Villarroya, F.; Pazienza, V.; et al. Efficacy and epigenetic interactions of novel DNA hypomethylating agent guadecitabine (SGI-110) in preclinical models of hepatocellular carcinoma. Epigenetics 2016. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Rappa, F.; Greco, A.; Podrini, C.; Cappello, F.; Foti, M.; Bourgoin, L.; Peyrou, M.; Marino, A.; Scibetta, N.; Williams, R.; et al. Immunopositivity for histone macroH2A1 isoforms marks steatosis-associated hepatocellular carcinoma. PLoS ONE 2013, 8, e54458. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.; Horvath, S.; Raj, K. Epigenetic clock analyses of cellular senescence and ageing. Oncotarget 2016, 7, 8524–8531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Sulli, G.; Di Micco, R.; d’Adda di Fagagna, F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Sporn, J.C.; Jung, B. Differential regulation and predictive potential of MacroH2A1 isoforms in colon cancer. Am. J. Pathol. 2012, 180, 2516–2526. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, M.; Ladurner, A.G. ATM, MacroH2A.1, and SASP: The checks and balances of cellular senescence. Mol. Cell 2015, 59, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Changolkar, L.N.; Costanzi, C.; Leu, N.A.; Chen, D.; McLaughlin, K.J.; Pehrson, J.R. Developmental changes in histone macroH2A1-mediated gene regulation. Mol. Cell Biol. 2007, 27, 2758–2764. [Google Scholar] [CrossRef] [PubMed]
- Changolkar, L.N.; Singh, G.; Cui, K.; Berletch, J.B.; Zhao, K.; Disteche, C.M.; Pehrson, J.R. Genome-wide distribution of macroH2A1 histone variants in mouse liver chromatin. Mol. Cell Biol. 2010, 30, 5473–5483. [Google Scholar]
- Buschbeck, M.; Uribesalgo, I.; Wibowo, I.; Rue, P.; Martin, D.; Gutierrez, A.; Morey, L.; Guigó, R.; López-Schier, H.; Di Croce, L. The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat. Struct. Mol. Biol. 2009, 16, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Borghesan, M.; Mazzoccoli, G.; Sheedfar, F.; Oben, J.; Pazienza, V.; Vinciguerra, M. Histone variants and lipid metabolism. Biochem. Soc. Trans. 2014, 42, 1409–1413. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Tryndyak, V.P.; Bagnyukova, T.V.; Melnyk, S.; Montgomery, B.; Ross, S.A.; Latendresse, J.R.; Rusyn, I.; Beland, F.A. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J. Hepatol. 2009, 51, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Pazienza, V.; Borghesan, M.; Mazza, T.; Sheedfar, F.; Panebianco, C.; Williams, R.; Mazzoccoli, G.; Andriulli, A.; Nakanishi, T.; Vinciguerra, M. SIRT1-metabolite binding histone macroH2A1.1 protects hepatocytes against lipid accumulation. Aging 2014, 6, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Podrini, C.; Koffas, A.; Chokshi, S.; Vinciguerra, M.; Lelliott, C.J.; White, J.K.; Adissu, H.A.; Williams, R.; Greco, A. MacroH2A1 isoforms are associated with epigenetic markers for activation of lipogenic genes in fat-induced steatosis. FASEB J. 2015, 29, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Boulard, M.; Storck, S.; Cong, R.; Pinto, R.; Delage, H.; Bouvet, P. Histone variant macroH2A1 deletion in mice causes female-specific steatosis. Epigenet. Chromat. 2010, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheedfar, F.; Vermeer, M.; Pazienza, V.; Villarroya, J.; Rappa, F.; Cappello, F.; Mazzoccoli, G.; Villarroya, F.; van der Molen, H.; Hofker, M.H.; et al. Genetic ablation of macrohistone H2A1 leads to increased leanness, glucose tolerance and energy expenditure in mice fed a high-fat diet. Int. J. Obes. 2015, 39, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marjanovic, M.P.; Hurtado-Bages, S.; Lassi, M.; Valero, V.; Malinverni, R.; Delage, H.; Navarro, M.; Corujo, D.; Guberovic, I.; Douet, J.; et al. MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD+ consumption. Nat. Struct. Mol. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dell’Orso, S.; Wang, A.H.; Shih, H.Y.; Saso, K.; Berghella, L.; Gutierrez-Cruz, G.; Ladurner, A.G.; O’Shea, J.J.; Sartorelli, V.; Zare, H. The histone variant MacroH2A1.2 is necessary for the activation of muscle enhancers and recruitment of the transcription factor Pbx1. Cell Rep. 2016, 14, 1156–1168. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Liu, C.; Sun, Y.; Wang, W.; Huang, K.; Zheng, L. MacroH2A1.1 cooperates with EZH2 to promote adipogenesis by regulating Wnt signaling. J. Mol. Cell Biol. 2017, 9, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Biase, S.; Longo, V.D. Fasting-induced differential stress sensitization in cancer treatment. Mol. Cell Oncol. 2016, 3, e1117701. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell 2014, 14, 810–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pehrson, J.R.; Changolkar, L.N.; Costanzi, C.; Leu, N.A. Mice without macroH2A histone variants. Mol. Cell Biol. 2014, 34, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Pasque, V.; Jullien, J.; Miyamoto, K.; Halley-Stott, R.P.; Gurdon, J.B. Epigenetic factors influencing resistance to nuclear reprogramming. Trends Genet. 2011, 27, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Gao, S.; Sung, L.Y.; Corry, G.N.; Ma, Y.; Nagy, Z.P.; Tian, X.C.; Rasmussen, T.P. Rapid elimination of the histone variant MacroH2A from somatic cell heterochromatin after nuclear transfer. Cell Reprogram. 2010, 12, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Pasque, V.; Radzisheuskaya, A.; Gillich, A.; Halley-Stott, R.P.; Panamarova, M.; Zernicka-Goetz, M.; Surani, M.A.; Silva, J.C. Histone variant macroH2A marks embryonic differentiation in vivo and acts as an epigenetic barrier to induced pluripotency. J. Cell Sci. 2012, 125 (Pt 24), 6094–6104. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M.J.; Sese, B.; Kuebler, B.; Bilic, J.; Boue, S.; Marti, M.; Izpisua Belmonte, J.C. Macrohistone variants preserve cell identity by preventing the gain of H3K4me2 during reprogramming to pluripotency. Cell Rep. 2013, 3, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M.J.; Sese, B.; Marti, M.; Izpisua Belmonte, J.C. Macro histone variants are critical for the differentiation of human pluripotent cells. J. Biol. Chem. 2013, 288, 16110–16116. [Google Scholar] [CrossRef] [PubMed]
- Gaspar-Maia, A.; Qadeer, Z.A.; Hasson, D.; Ratnakumar, K.; Leu, N.A.; Leroy, G.; Liu, S.; Costanzi, C.; Valle-Garcia, D.; Schaniel, C.; et al. MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat. Commun. 2013, 4, 1565. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lv, P.; Yan, G.; Fan, H.; Cheng, L.; Zhang, F.; Dang, Y.; Wu, H.; Wen, B. MacroH2A1 associates with nuclear lamina and maintains chromatin architecture in mouse liver cells. Sci. Rep. 2015, 5, 17186. [Google Scholar] [CrossRef] [PubMed]
- Douet, J.; Corujo, D.; Malinverni, R.; Renauld, J.; Sansoni, V.; Marjanovic, M.P.; Cantari’o, N.; Valero, V.; Mongelard, F.; Bouvet, P.; et al. MacroH2A histone variants maintain nuclear organization and heterochromatin architecture. J. Cell Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Angelov, D.; Molla, A.; Perche, P.Y.; Hans, F.; Cote, J.; Khochbin, S.; Bouvet, P.; Dimitrov, S. The histone variant macroH2A interferes with transcription factor binding and SWI/SNF nucleosome remodeling. Mol. Cell 2003, 11, 1033–1041. [Google Scholar] [CrossRef]
- Ratnakumar, K.; Duarte, L.F.; LeRoy, G.; Hasson, D.; Smeets, D.; Vardabasso, C.; Bonisch, C.; Zeng, T.; Xiang, B.; Zhang, D.Y.; et al. ATRX-mediated chromatin association of histone variant macroH2A1 regulates alpha-globin expression. Genes Dev. 2012, 26, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, P.V.; Ahel, D.; Ryan, D.P.; Weston, R.; Wiechens, N.; Kraehenbuehl, R.; Owen-Hughes, T.; Ahel, I. DNA repair factor APLF is a histone chaperone. Mol. Cell 2011, 41, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Hussey, K.M.; Chen, H.; Yang, C.; Park, E.; Hah, N.; Erdjument-Bromage, H.; Tempst, P.; Gamble, M.J.; Kraus, W.L. The histone variant MacroH2A1 regulates target gene expression in part by recruiting the transcriptional coregulator PELP1. Mol. Cell Biol. 2014, 34, 2437–2449. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; Novikov, L.; Casill, A.D.; Park, J.W.; Gamble, M.J. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat. Struct. Mol. Biol. 2014, 21, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Jufvas, A.; Stralfors, P.; Vener, A.V. Histone variants and their post-translational modifications in primary human fat cells. PLoS ONE 2011, 6, e15960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, F.; Nusinow, D.A.; Chalkley, R.J.; Plath, K.; Panning, B.; Burlingame, A.L. Mapping post-translational modifications of the histone variant MacroH2A1 using tandem mass spectrometry. Mol. Cell Proteom. 2006, 5, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Muratore-Schroeder, T.L.; Diaz, R.L.; Chow, J.C.; Changolkar, L.N.; Shabanowitz, J.; Heard, E.; Pehrson, J.R.; Hunt, D.F.; Allis, C.D. A phosphorylated subpopulation of the histone variant macroH2A1 is excluded from the inactive X chromosome and enriched during mitosis. Proc. Natl. Acad. Sci. USA 2008, 105, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.; Scheffzek, K.; et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Sellou, H.; Lebeaupin, T.; Chapuis, C.; Smith, R.; Hegele, A.; Singh, H.R.; Kozlowski, M.; Bultmann, S.; Ladurner, A.G.; Timinszky, G.; et al. The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage. Mol. Biol. Cell 2016, 27, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Natoli, G. Control of NF-κB-dependent transcriptional responses by chromatin organization. Cold Spring Harb. Perspect. Biol. 2009, 1, a000224. [Google Scholar] [CrossRef] [PubMed]
- Takase, O.; Yoshikawa, M.; Idei, M.; Hirahashi, J.; Fujita, T.; Takato, T.; Isagawa, T.; Nagae, G.; Suemori, H.; Aburatani, H.; et al. The role of NF-κB signaling in the maintenance of pluripotency of human induced pluripotent stem cells. PLoS ONE 2013, 8, e56399. [Google Scholar] [CrossRef] [PubMed]
- Michael, S.; Achilleos, C.; Panayiotou, T.; Strati, K. Inflammation shapes stem cells and stemness during infection and beyond. Front. Cell Dev. Biol. 2016, 4, 118. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Li, Y.; Bhattacharya, S.; O’Connor, M.; Pu, C.; Lin, J.; Wang, T.; Xiang, D.; Kong, L.; Wei, M.Q.; et al. Inflammation and cancer stem cells. Cancer Lett. 2014, 345, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Arany, P.R.; Cho, A.; Hunt, T.D.; Sidhu, G.; Shin, K.; Hahm, E.; Huang, G.X.; Weaver, J.; Chen, A.C.; Padwa, B.L.; et al. Photoactivation of endogenous latent transforming growth factor-β1 directs dental stem cell differentiation for regeneration. Sci. Transl. Med. 2014, 6, 238ra69. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Chen, W.Y.; Li, H.Y.; Chien, Y.; Chang, W.C.; Hsieh, P.C.; Wu, P.; Chen, C.Y.; Song, H.Y.; Chien, C.S.; et al. CHD1L Regulated PARP1-driven pluripotency and chromatin remodeling during the early-stage cell reprogramming. Stem Cells 2015, 33, 2961–2972. [Google Scholar] [CrossRef] [PubMed]
- Ouararhni, K.; Hadj-Slimane, R.; Ait-Si-Ali, S.; Robin, P.; Mietton, F.; Harel-Bellan, A.; Dimitrov, S.; Hamiche, A. The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Genes Dev. 2006, 20, 3324–3336. [Google Scholar] [CrossRef] [PubMed]
- Ladurner, A.; Sporn, J.; Muley, T. Diagnostic Method for Predicting the Risk of Cancer Recurrence Based on Histone MacroH2A Isoforms. Google Patents CA2766656 A1, 1 Januray 2010. [Google Scholar]
- Hamiche, A. MacroH2A Non-Histone Domain as Inhibitor of PARP-1 Activity and Uses Thereof. Google Patents EP1948215 B1, 11 January 2012. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Histone variant | Species | Process/ Disease | Tissue/Cells | Up/Down-Regulation | Reference |
|---|---|---|---|---|---|
| MacroH2A | mouse | NASH | liver | Up-regulation | [39] |
| MacroH2A1.1 | mouse | NASH/HCC | liver | Up-regulation | [27] |
| MacroH2A1.2 | mouse | NASH/HCC | liver | Up-regulation | [27] |
| MacroH2A1.1 | human | NAFLD | liver | Up-regulation | [27] |
| MacroH2A1.2 | human | NAFLD/HCC | liver | Up-regulation | [27] |
| MacroH2A1.2 | human | steatosis | HepG2/IHHs | Up-regulation | [41] |
| MacroH2A1.1 | mouse | steatosis | liver | Up-regulation | [41] |
| MacroH2A1.1 | human | obesity | adipose tissue | Up-regulation | [14] |
| MacroH2A1.2 | human | obesity | adipose tissue | Down-regulation | [14] |
| MacroH2A1.1 | mouse | obesity | adipose tissue | Up-regulation | [48] |
| Histone variant | Model | Overexpression | Phenotype | Reference |
|---|---|---|---|---|
| MacroH2A1 | mouse | KD | Glucose intolerance, increased hepatic lipidogenic gene expression | [42] |
| MacroH2A1 | mouse | KD | Fatty liver in 50% of females; overexpression of the X-linked thyroxine-binding globuline gene | [44] |
| MacroH2A1.1 | Hepatoma cells | OE | Antilipidogenic | [40] |
| MacroH2A1.2 | Hepatoma cells | OE | Prolidipogenic | [40] |
| MacroH2A1.1 | 3T3-L1 | OE | Proadipogenic | [14] |
| MacroH2A1.2 | 3T3-L1 | OE | Antiadipogenic | [14] |
| MacroH2A1.1 | 3T3-L1 | OE | Proadipogenic | [48] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Re, O.; Vinciguerra, M. Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration. Genes 2017, 8, 367. https://doi.org/10.3390/genes8120367
Lo Re O, Vinciguerra M. Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration. Genes. 2017; 8(12):367. https://doi.org/10.3390/genes8120367
Chicago/Turabian StyleLo Re, Oriana, and Manlio Vinciguerra. 2017. "Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration" Genes 8, no. 12: 367. https://doi.org/10.3390/genes8120367
APA StyleLo Re, O., & Vinciguerra, M. (2017). Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration. Genes, 8(12), 367. https://doi.org/10.3390/genes8120367