Linking DNA Damage and Age-Related Promoter DNA Hyper-Methylation in the Intestine


Abstract
:1. Introduction
2. Methods
2.1. 3D Multiscale Model of Epigenetic Aging of the Intestinal Epithelium
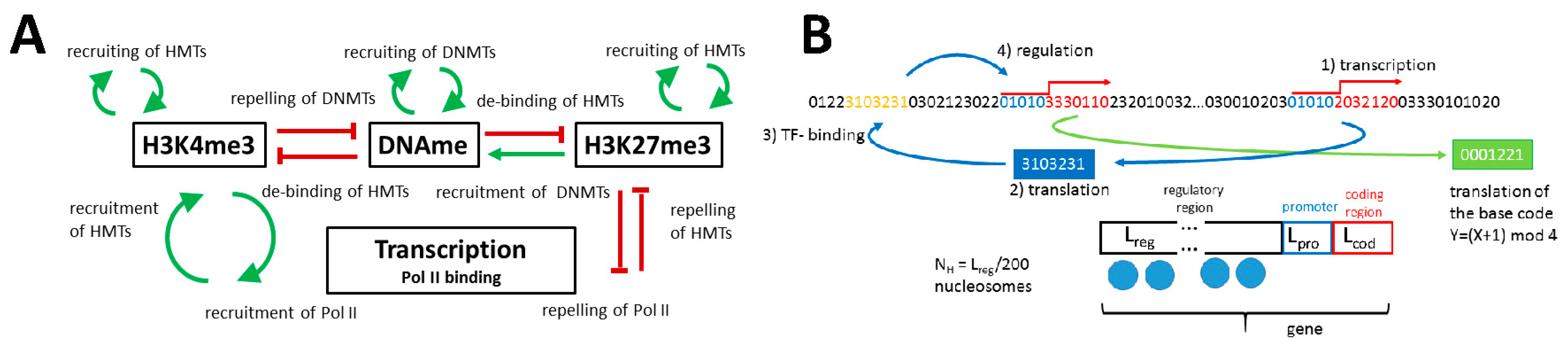
2.1.1. The Model of Epigenetic Regulation of Transcription
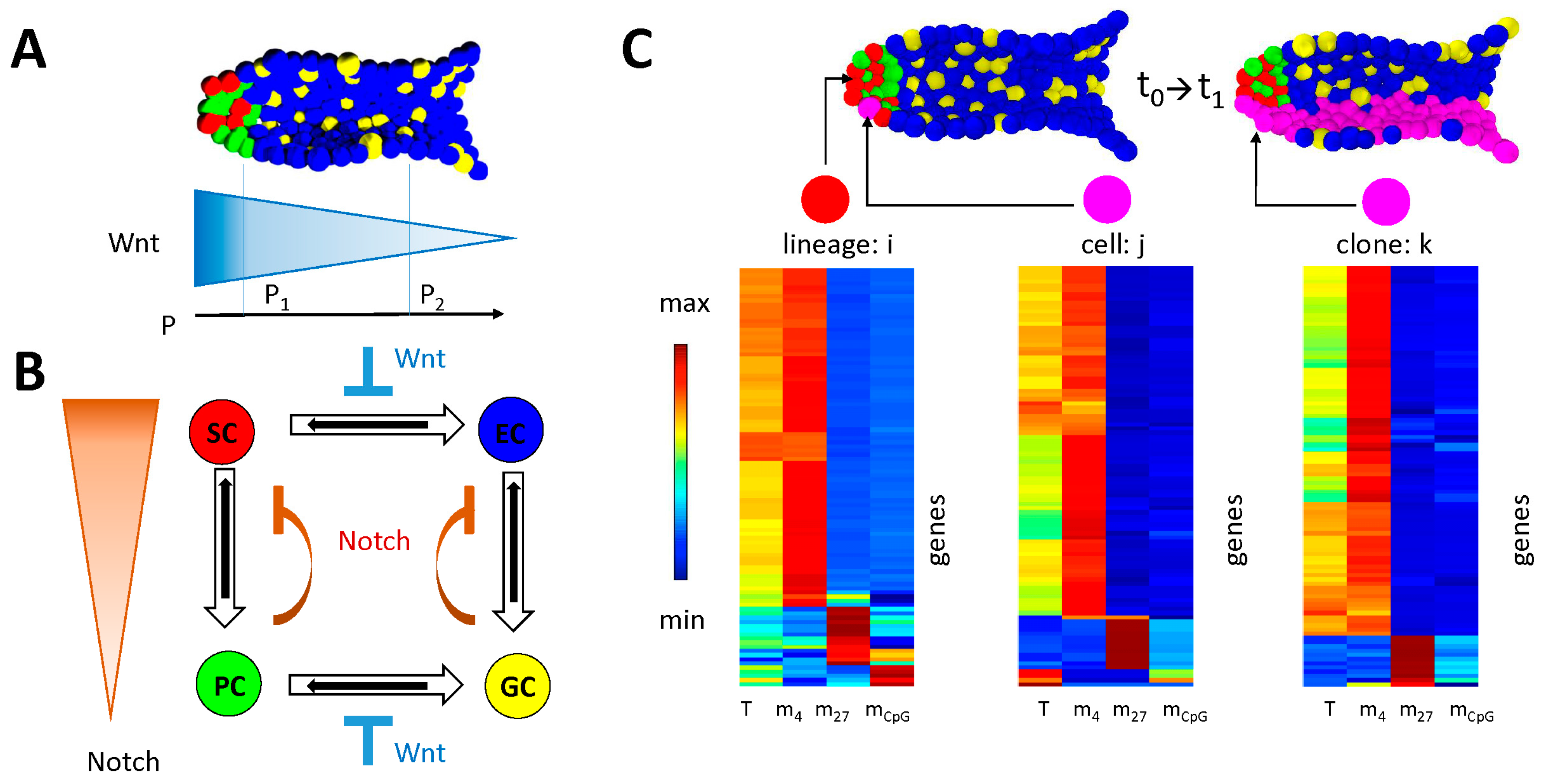
2.1.2. The 3D Individual Cell-Based Model of the Intestinal Crypt
2.2. Model of DNA Damage and Damage Repair in the Crypt
3. Results
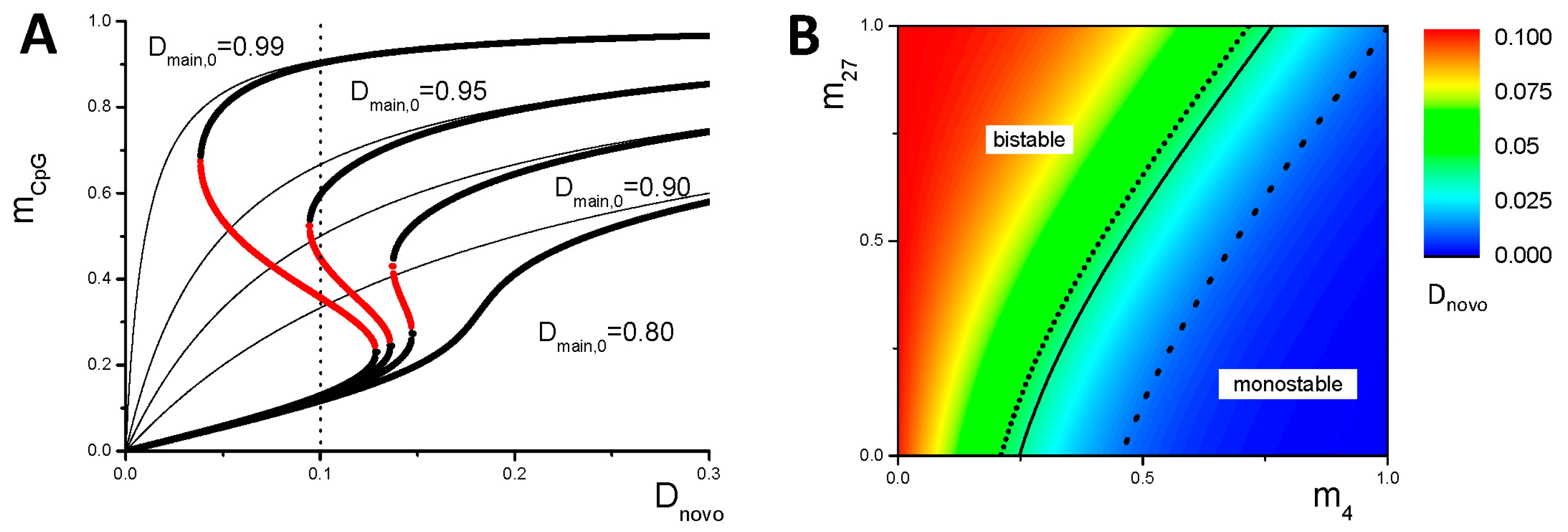
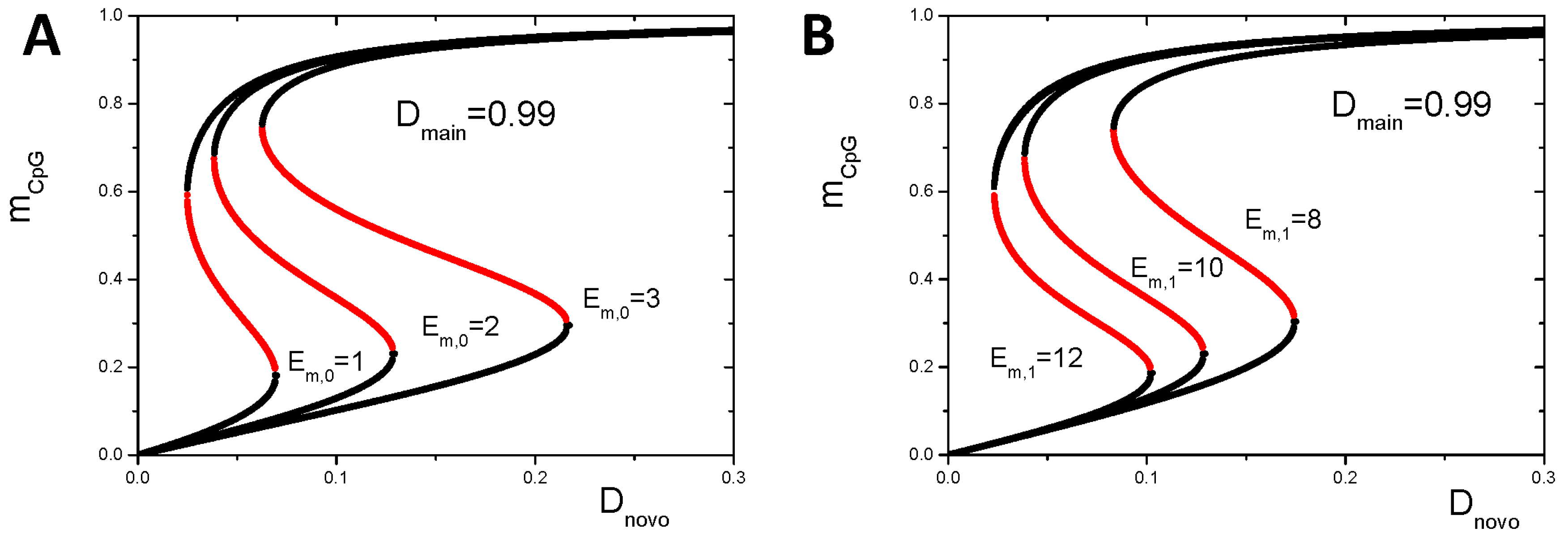
3.1. Maintenance of DNA Methylation with Positive Auto-Feedback
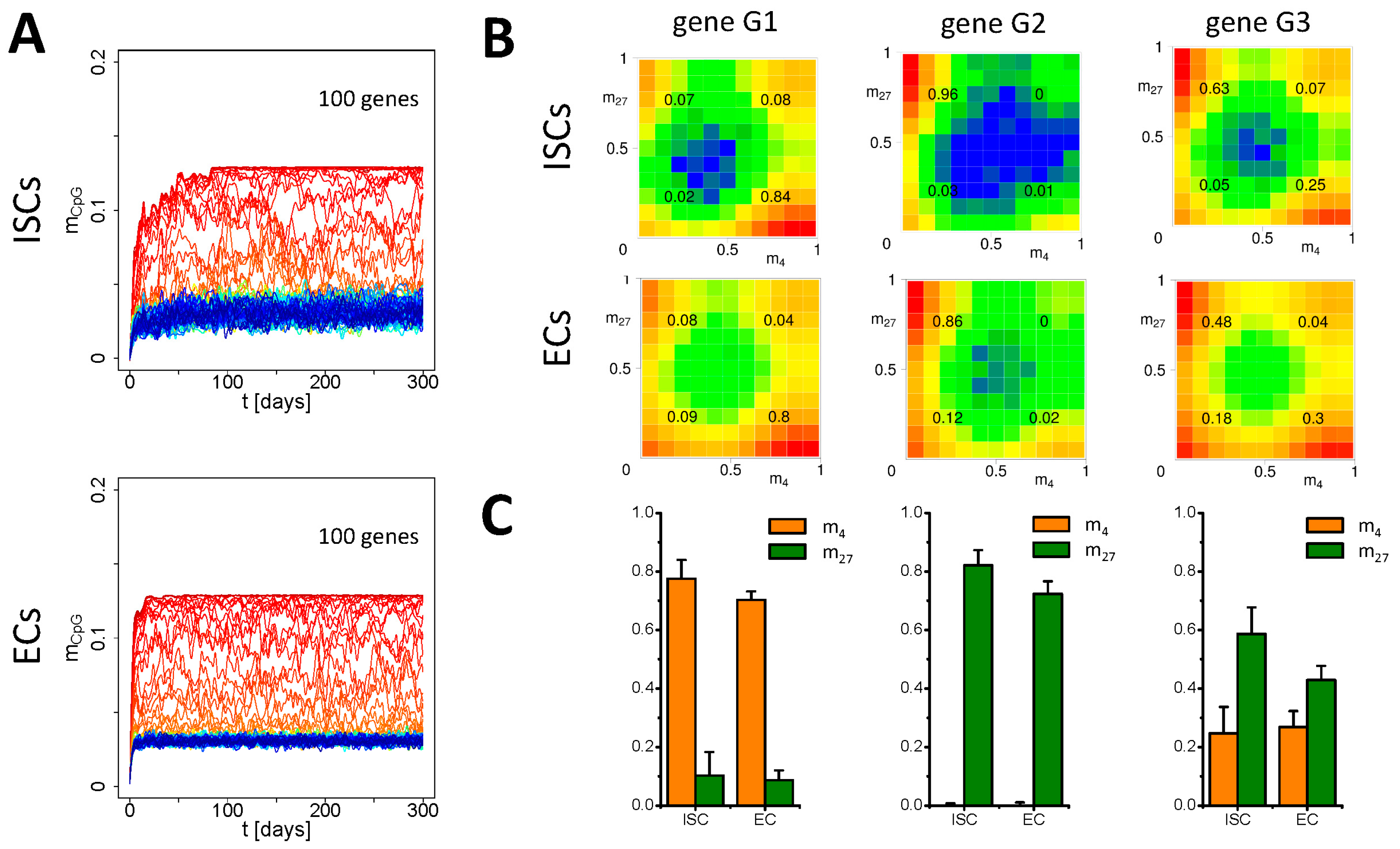
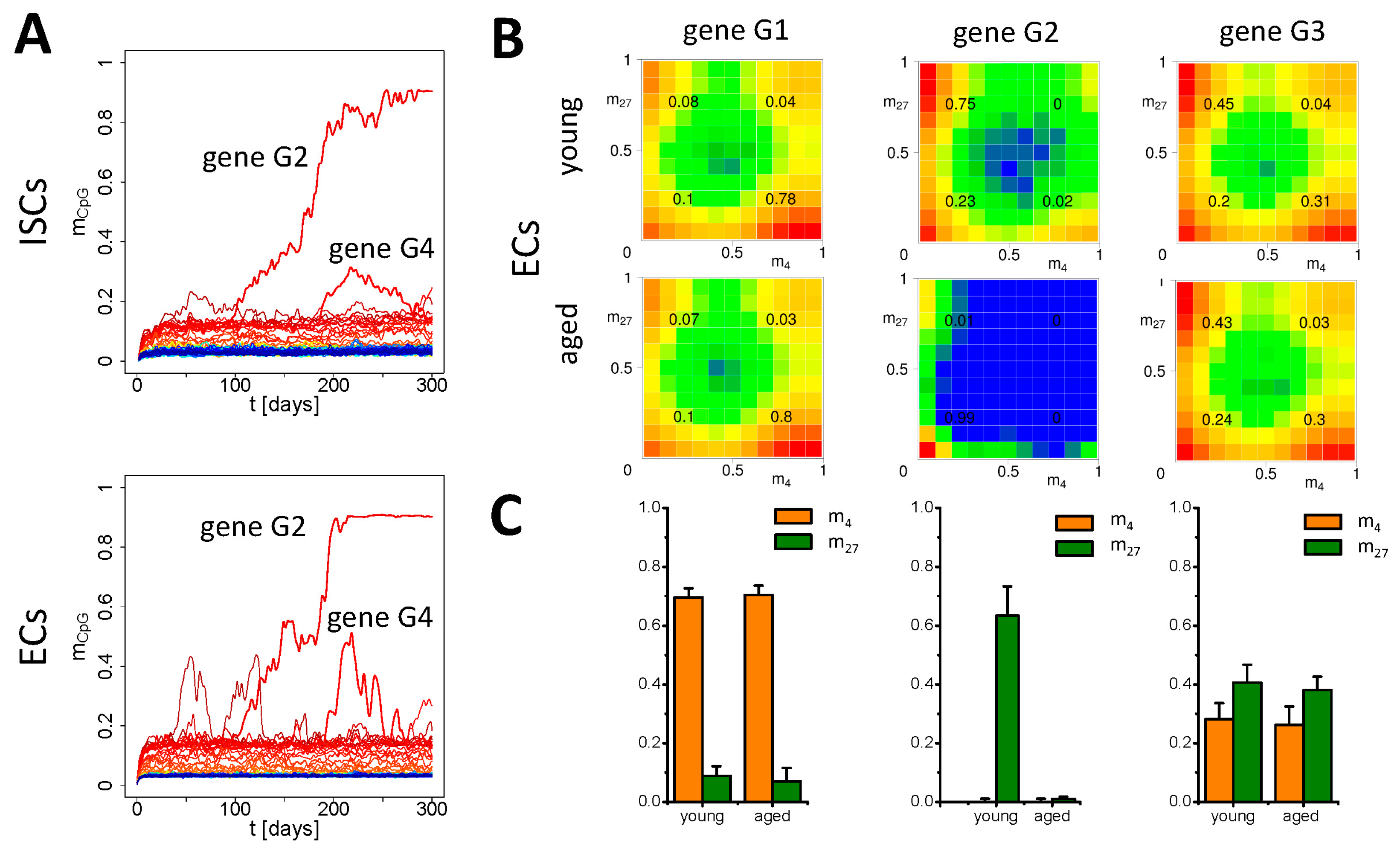
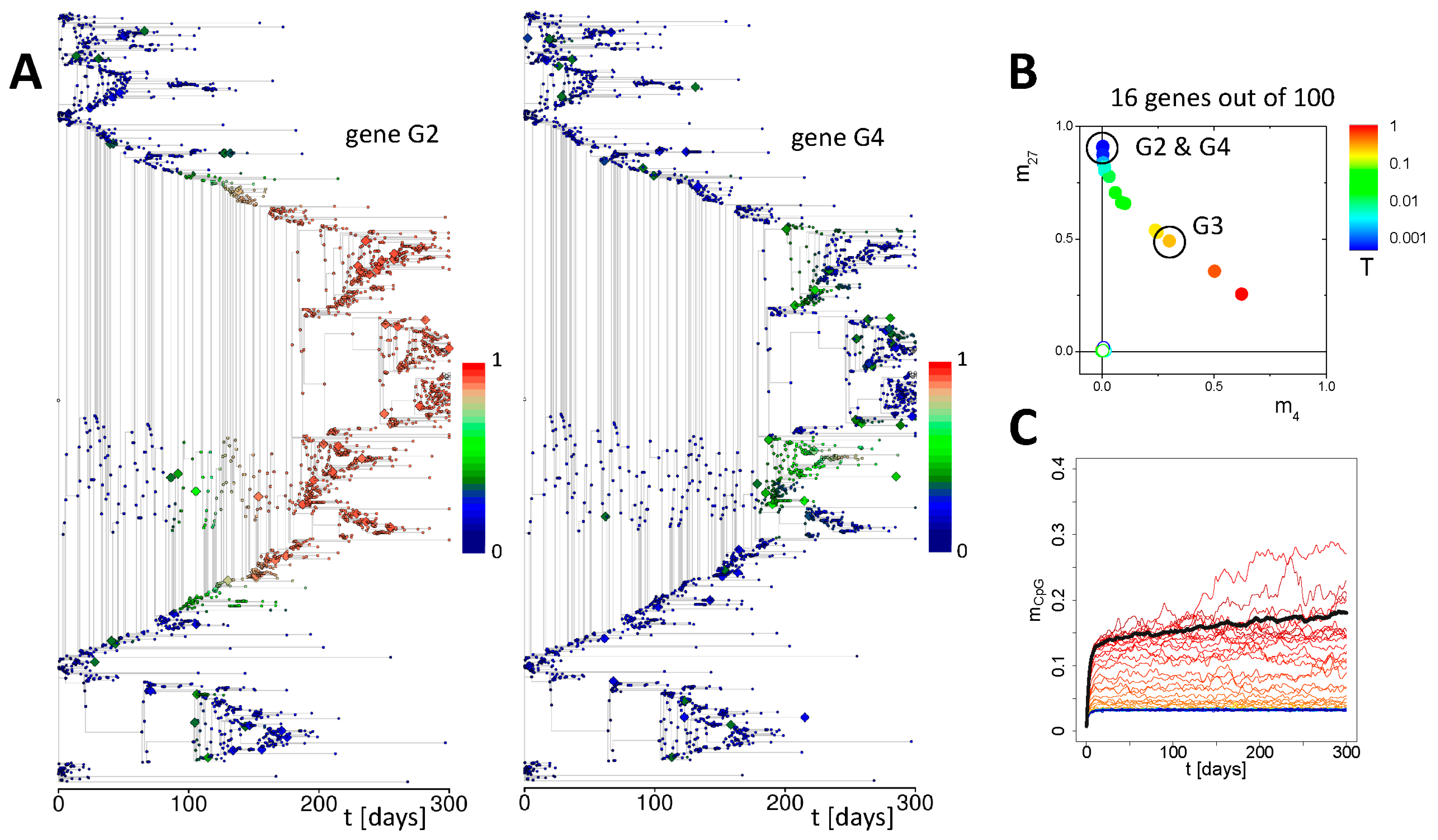
3.2. Simulation of Aging without Damage
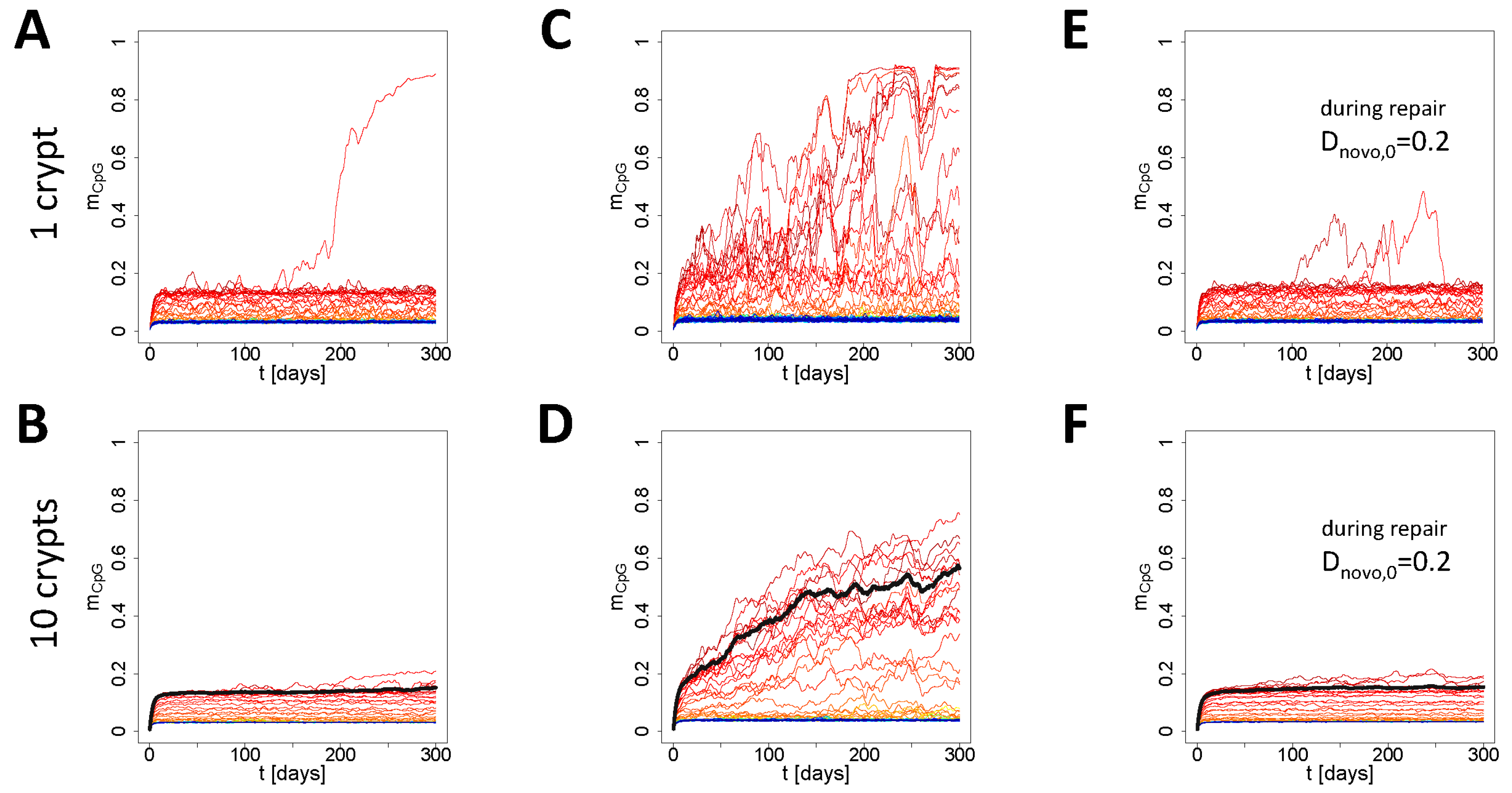
3.3. Simulation of Aging under Repeated DDR
4. Discussion
Acknowledgments
Author contributions
Conflicts of Interest
Abbreviations
| ISC | Intestinal stem cell |
| PC | Paneth cell |
| GC | Goblet cell |
| EC | Enterocyte |
| DSB | Double-strand break |
| DDR | DNA damage repair |
| HDM | Histone demethylases |
| HMT | Histone methyltransferase |
| DNMT | DNA methyltransferase |
Appendix A. Model Description
Appendix A1. Model of the Epigenetic Regulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Description |
|---|---|---|
| Histone modification | ||
| , | 10.0, 11.0 | ground enthalpy per bound HMT * |
| , | −5.0, −5.0 | free energy of CpG binding * |
| , | −0.8, −0.8 | free energy of histone binding * |
| , | 0.1, 0.1 | modification constant |
| , | 0.01 (0), 0.01 | de-modification constant |
| NH | 10 | number of cooperative nucleosomes |
| DNA methylation | ||
| Dmain,0 | 0.99 | probability of maintaining DNA methylation |
| Dnovo,0 | 0.1 (0.3) | probability of de novo DNA methylation |
| , | 6, 4 | interaction energy between HMTs and DNMTs * |
| Em,0 | 2 | ground enthalpy per bound maintenance DNMT * |
| Em,1 | −10 | free energy of methyl-CG binding * |
Appendix A2. Model of Transcriptional Regulation
| Parameter | Value | Description |
|---|---|---|
| Pmax | 100 | maximum promoter activity |
| δ | 0.1 | transcript degradation constant |
| εA | 1/−1 | free energy change of polymerase binding for activators/repressors * |
| Lreg | 2.000 bp | length of the regulatory region |
| Lpro | 5 bp | length of the promoter region |
| Lcod | 7 bp | length of the coding region |
Appendix A3. Crypt Model
| Symbol | Value | Parameter | References |
|---|---|---|---|
| Parameter of the cell model | |||
| V0 | 4/3π (5 µm)3 | minimal volume of an isolated cell | estimated |
| τ | 14 h | cell growth time | results in an effective cell cycle time ~24 h |
| E | 1 kPa | Young modulus | [51] |
| ν | 1/3 | Poisson ratio | |
| εc | 200 µN/m | cell-cell anchorage | |
| Vp | 0.88 V0 | threshold volume for contact inhibition | set |
| Parameter of the Basal Membrane model | |||
| z0 | 150 µm | length of the crypt | set, according to properties of the crypt shape [20] |
| r0 | 60 µm | crypt radius at the crypt-villus junction | |
| λ1 | 0.25 | shape parameter 1 | |
| λ2 | 0.1 | shape parameter 2 | |
| Ω | 0.95 | optimal cell-BM distance in cell radii | set |
| 35 10−12 Nm | maximum cell-knot interaction energy of PCs | set | |
| 5.5 10−12 Nm | maximum cell-knot interaction energy of all other cells | ensuring apoptosis rates < 5% [52] | |
| Parameter of crypt dynamics | |||
| ηc | 5 1010 Ns/m3 | friction constant for cell-cell friction | [51] |
| ηBM | 3.2 Ns/m | friction coefficient for cell-BM friction | fit: turnover |
| ηVO | 400 Ns/m | friction coefficient regarding volume changes | [51] |
| 7.5 nN | active migration force of PCs | fit: distribution of PCs | |
| 4.5 nN | active migration force of all other cells | fit: turnover and Brdu data | |
| Parameter of the lineage specification and differentiation model | |||
| P1 | −129 µm | position of the Wnt- threshold TPWnt for priming | fit: size of the PC compartment |
| P2 | −87.5 µm | position of the Wnt- threshold TDWnt for differentiation | fit: turnover and Brdu data |
| N1 | 3 | minimum number of Notch-ligand expressing neighbors in the niche | set: refers to TDNotch /LPPaneth and TDNotch/LPGoblet [20] |
| N2 | 1 | minimum number of Notch-ligand expressing neighbors outside the niche | |
| τP | 8 weeks | average PC lifespan with contact to SCs | [19] |
| τA | 12 h | max. PC lifespan without contact to SCs | |
Appendix B. Additional Simulation Results
Appendix B1. Bistable DNA Methylation States

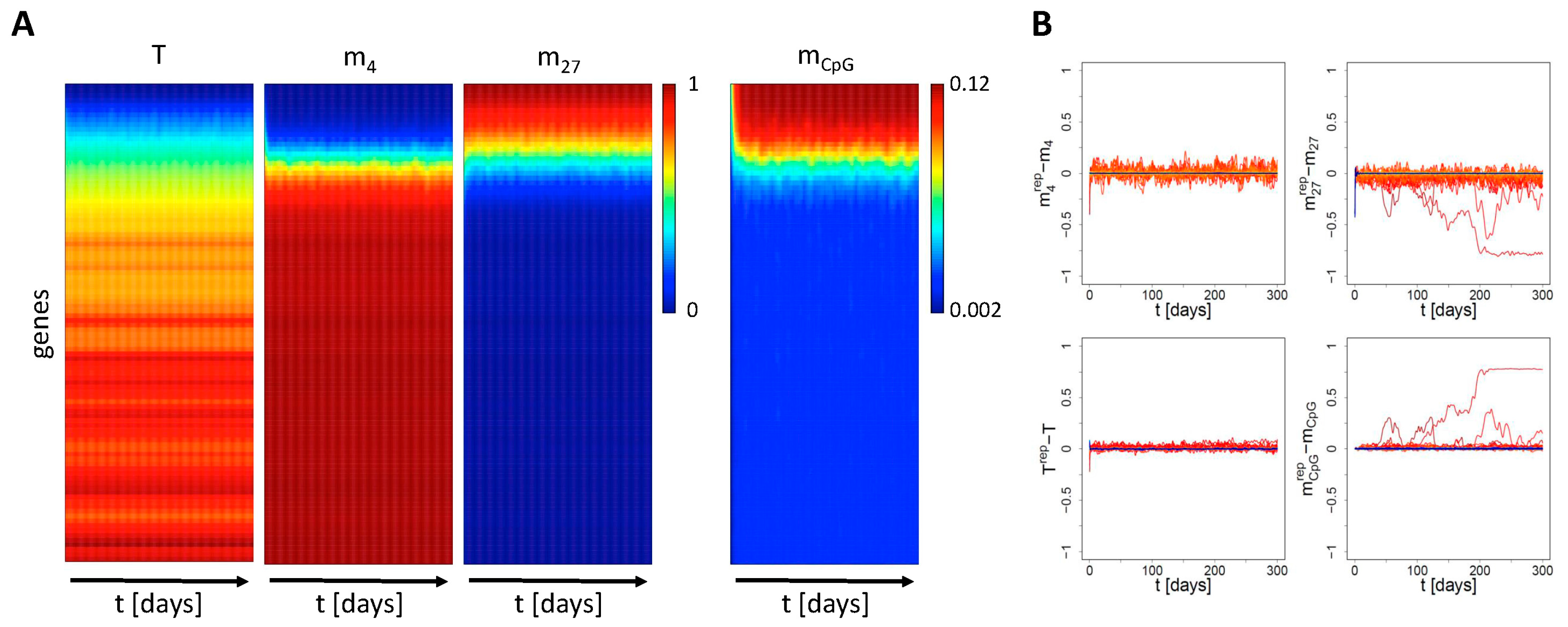
Appendix B2. Simulation of Aging


References
- Christensen, B.C.; Houseman, E.A.; Marsit, C.J.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Ritz, B.R. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging 2015, 7, 1130. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Levine, A.J. HIV-1 infection accelerates age according to the epigenetic clock. J. Infect. Dis. 2015, 212, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P.; et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, J.; Galle, J.; Rohlf, T. Is adult stem cell aging driven by conflicting modes of chromatin remodeling? Bioessays 2012, 34, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, J.; Rohlf, T.; Loeffler, M.; Galle, J. Understanding epigenetic changes in aging stem cells—A computational model approach. Aging Cell 2014, 13, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, S.; Hinkal, G.; Kim, H.S.; Shen, L.; Zhang, L.; Zhang, J.; Zhang, N.; Liang, S.; Donehower, L.A.; Issa, J.P. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Noreen, F.; Röösli, M.; Gaj, P.; Pietrzak, J.; Weis, S.; Urfer, P.; Regula, J.; Schär, P.; Truninger, K. Modulation of age- and cancer-associated DNA methylation change in the healthy colon by aspirin and lifestyle. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Ikegami, D.; Wakabayashi, M.; Niwa, T.; Kim, Y.J.; Ushijima, T. Induction of aberrant trimethylation of histone H3 lysine 27 by inflammation in mouse colonic epithelial cells. Carcinogenesis 2012, 33, 2384–2390. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Coelho, R.; Grácio, D.; Dias, C.; Silva, M.; Peixoto, A.; Lopes, P.; Costa, C.; Teixeira, J.P.; Macedo, G.; et al. DNA damage and oxidative DNA damage in inflammatory bowel disease. J. Crohns Colitis 2016, 10, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Gursoy-Yuzugullu, O.; House, N.; Price, B.D. Patching Broken DNA: Nucleosome Dynamics and the Repair of DNA Breaks. J. Mol. Biol. 2016, 428, 1846–1860. [Google Scholar] [CrossRef] [PubMed]
- Thalheim, T.; Buske, P.; Przybilla, J.; Rother, K.; Loeffler, M.; Galle, J. Stem cell competition in the gut: Insights from multi-scale computational modelling. J. R. Soc. Interface 2016, 13, 20160218. [Google Scholar] [CrossRef] [PubMed]
- Buske, P.; Galle, J.; Barker, N.; Aust, G.; Clevers, H.; Loeffler, M. A comprehensive model of the spatio-temporal stem cell and tissue organisation in the intestinal crypt. PLoS Comput. Biol. 2011, 7, e1001045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thalheim, T.; Herberg, M.; Loeffler, M.; Galle, J. The Regulatory Capacity of Bivalent Genes-A Theoretical Approach. Int. J. Mol. Sci. 2017, 18, 1069. [Google Scholar] [CrossRef] [PubMed]
- Buratowski, S.; Kim, T. The Role of Cotranscriptional Histone Methylations. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2010; Volume 75, pp. 95–102. [Google Scholar]
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pijnappel, W.W.; van Schaik, F.M.; Varier, R.A.; Baltissen, M.P.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 2007, 131, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Jermann, P.; Hoerner, L.; Burger, L.; Schübeler, D. Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. PNAS 2014, 111, E3415–E3421. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Malatesta, M.; Jung, H.R.; Walfridsson, J.; Willer, A.; Olsson, L.; Skotte, J.; Wutz, A.; Porse, B.; Jensen, O.N.; et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 2010, 38, 4958–4969. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Barth, T.K.; Reverón-Gómez, N.; Sidoli, S.; Schmidt, A.; Jensen, O.N.; Imhof, A.; Groth, A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 2015, 29, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.P.; Skene, P.J.; Selfridge, J.; Clouaire, T.; Guy, J.; Webb, S.; Kerr, A.R.; Deaton, A.; Andrews, R.; James, K.D.; et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 2010, 464, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Rush, M.; Appanah, R.; Lee, S.; Lam, L.L.; Goyal, P.; Lorincz, M.C. Targeting of EZH2 to a defined genomic site is sufficient for recruitment of Dnmt3a but not de novo DNA methylation. Epigenetics 2009, 4, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Wirth, H.; Galle, J. Gene expression density profiles characterize modes of genomic regulation: Theory and experiment. J. Biotechnol. 2010, 149, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Qian, X.; Shen, J.; Wang, Y.; Li, X.; Liu, R.; Xia, Y.; Chen, Q.; Peng, G.; Lin, S.Y.; Lu, Z. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat. Cell Biol. 2015, 17, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Cloos, P.A.; Christensen, J.; Agger, K.; Helin, K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008, 22, 1115–1140. [Google Scholar] [CrossRef] [PubMed]
- Boulard, M.; Edwards, J.R.; Bestor, T.H. FBXL10 protects Polycomb-bound genes from hypermethylation. Nat. Genet. 2015, 47, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Sontag, L.B.; Lorincz, M.C.; Luebeck, E.G. Dynamics, stability and inheritance of somatic DNA methylation imprints. J. Theor. Biol. 2006, 242, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Sormani, G.; Haerter, J.O.; Lövkvist, C.; Sneppen, K. Stabilization of epigenetic states of CpG islands by local cooperation. Mol. Biosyst. 2016, 12, 2142–2146. [Google Scholar] [CrossRef] [PubMed]
- Luebeck, E.G.; Curtius, K.; Hazelton, W.D.; Maden, S.; Yu, M.; Thota, P.N.; Patil, D.T.; Chak, A.; Willis, J.E.; Grady, W.M. Identification of a key role of widespread epigenetic drift in Barrett’s esophagus and esophageal adenocarcinoma. Clin. Epigenetics 2017, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Laget, S.; Miotto, B.; Chin, H.G.; Estève, P.O.; Roberts, R.J.; Pradhan, S.; Defossez, P.A. MBD4 cooperates with DNMT1 to mediate methyl-DNA repression and protects mammalian cells from oxidative stress. Epigenetics 2014, 9, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Li, M.; Wu, H.; Jia, S.; Zhang, N.; Dai, Y.; Zhao, M.; Lu, Q. Down-regulation of MBD4 contributes to hypomethylation and overexpression of CD70 in CD4+ T cells in systemic lupus erythematosus. Clin. Epigenetics 2017, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Kazakevych, J.; Sayols, S.; Messner, B.; Krienke, C.; Soshnikova, N. Dynamic changes in chromatin states during specification and differentiation of adult intestinal stem cells. Nucleic Acids Res. 2017, 45, 5770–5784. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Revell, L.J. Phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet 2008, 4, e1000155. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, J.; Hopp, L.; Lübbert, M.; Loeffler, M.; Galle, J. Targeting DNA hypermethylation: Computational modeling of DNA demethylation treatment of acute myeloid leukemia. Epigenetics 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Franzen, J.; Zirkel, A.; Blake, J.; Rath, B.; Benes, V.; Papantonis, A.; Wagner, W. Senescence-associated DNA methylation is stochastically acquired in subpopulations of mesenchymal stem cells. Aging Cell 2017, 16, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Teodoridis, J.M.; Hardie, C.; Brown, R. CpG island methylator phenotype (CIMP) in cancer: Causes and implications. Cancer Lett. 2008, 268, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Remely, M.; Ferk, F.; Sterneder, S.; Setayesh, T.; Kepcija, T.; Roth, S.; Noorizadeh, R.; Greunz, M.; Rebhan, I.; Wagner, K.H.; et al. Vitamin E modifies high-fat diet-induced increase of DNA strand breaks, and changes in expression and DNA methylation of Dnmt1 and MLH1 in C57BL/6J Male Mice. Nutrients 2017, 9, 607. [Google Scholar] [CrossRef] [PubMed]
- Janzer, A.; Stamm, K.; Becker, A.; Zimmer, A.; Buettner, R.; Kirfel, J. The H3K4me3 histone demethylase Fbxl10 is a regulator of chemokine expression, cellular morphology, and the metabolome of fibroblasts. J. Biol. Chem. 2012, 287, 30984–30992. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Steiner, L.; Przybilla, J.; Rohlf, T.; Prohaska, S.; Galle, J. Transcriptional regulation by histone modifications: Towards a theory of chromatin re-organization during stem cell differentiation. Phys. Biol. 2013, 10, 026006. [Google Scholar] [CrossRef] [PubMed]
- Galle, J.; Loeffler, M.; Drasdo, D. Modeling the effect of deregulated proliferation and apoptosis on the growth dynamics of epithelial cell populations in vitro. Biophys. J. 2005, 88, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Marshman, E.; Ottewell, P.D.; Potten, C.S.; Watson, A.J. Caspase activation during spontaneous and radiation-induced apoptosis in the murine intestine. J. Pathol. 2001, 195, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Reinhardt, R.; Jeltsch, A. Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res. 2006, 34, 1182–1188. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thalheim, T.; Herberg, M.; Galle, J. Linking DNA Damage and Age-Related Promoter DNA Hyper-Methylation in the Intestine. Genes 2018, 9, 17. https://doi.org/10.3390/genes9010017
Thalheim T, Herberg M, Galle J. Linking DNA Damage and Age-Related Promoter DNA Hyper-Methylation in the Intestine. Genes. 2018; 9(1):17. https://doi.org/10.3390/genes9010017
Chicago/Turabian StyleThalheim, Torsten, Maria Herberg, and Joerg Galle. 2018. "Linking DNA Damage and Age-Related Promoter DNA Hyper-Methylation in the Intestine" Genes 9, no. 1: 17. https://doi.org/10.3390/genes9010017
APA StyleThalheim, T., Herberg, M., & Galle, J. (2018). Linking DNA Damage and Age-Related Promoter DNA Hyper-Methylation in the Intestine. Genes, 9(1), 17. https://doi.org/10.3390/genes9010017