Comprehensive Profiling of lincRNAs in Lung Adenocarcinoma of Never Smokers Reveals Their Roles in Cancer Development and Prognosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Never-Smoker Patients with Lung Adenocarcinoma
2.2. RNA-Sequencing Data and Long Intergenic Non-Coding RNA Detection
2.3. Long Intergenic Non-Coding RNA Differential Expression between Tumors and Normal Lungs
2.4. Long Intergenic Non-Coding RNA Association with Clinical Variables and Patient Survival
2.5. Validation Dataset from the Cancer Genome Atlas
3. Results
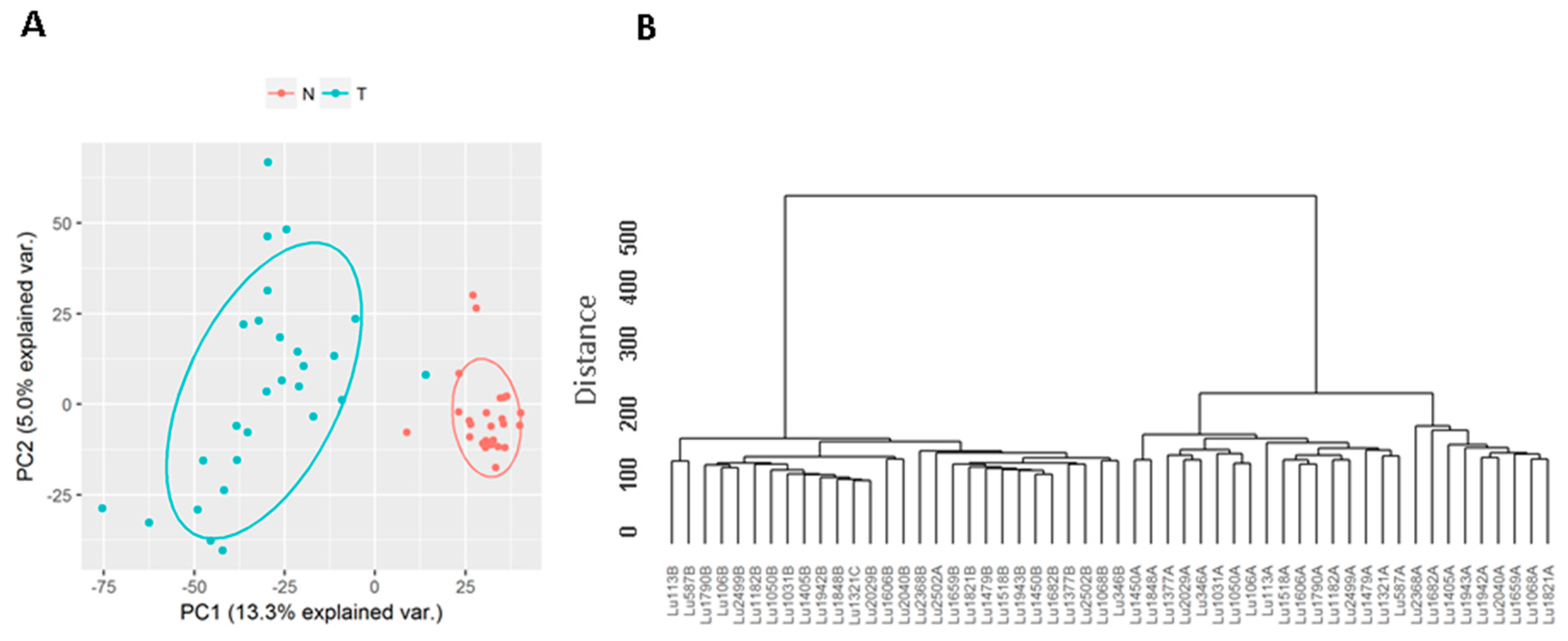
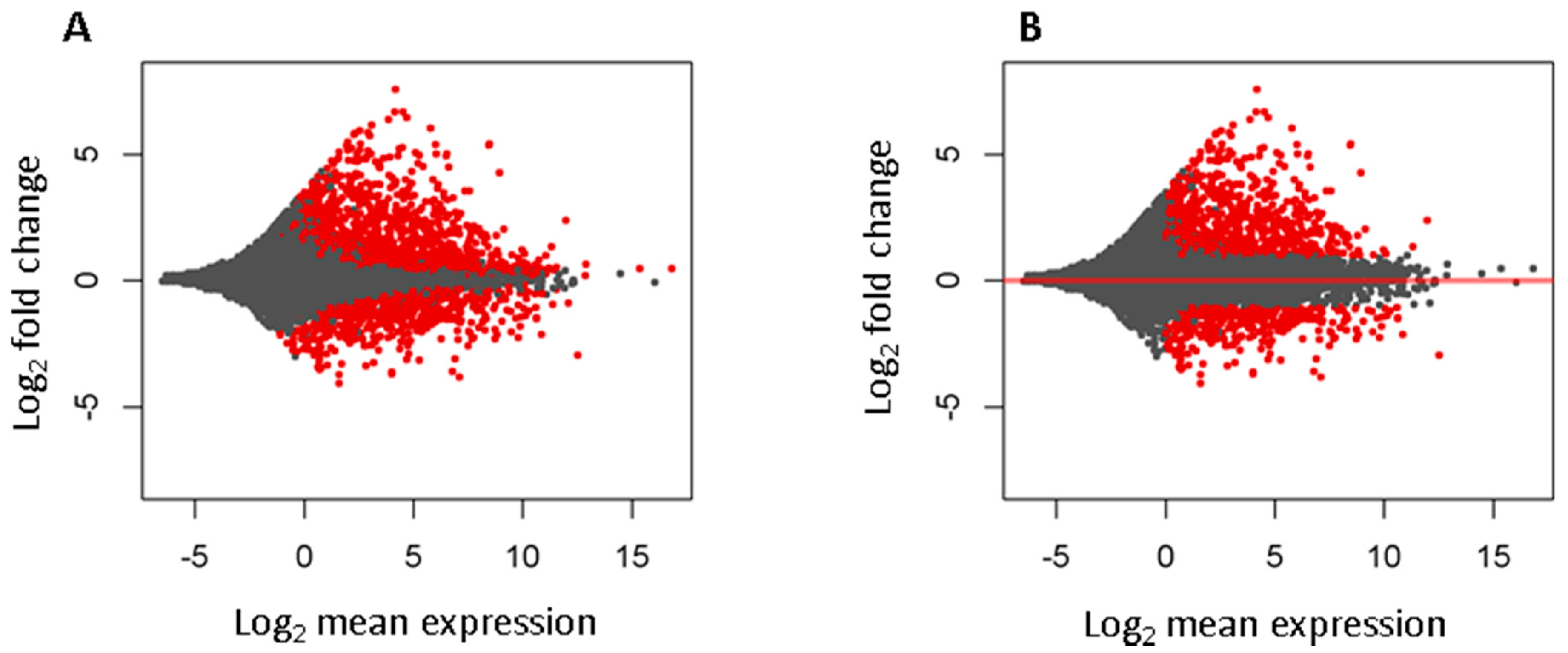
3.1. Known Long Intergenic Non-Coding RNA Expression between Tumor and Normal Lung Samples
3.2. Long Intergenic Non-Coding RNA and Potential Target Protein Coding Gene Association
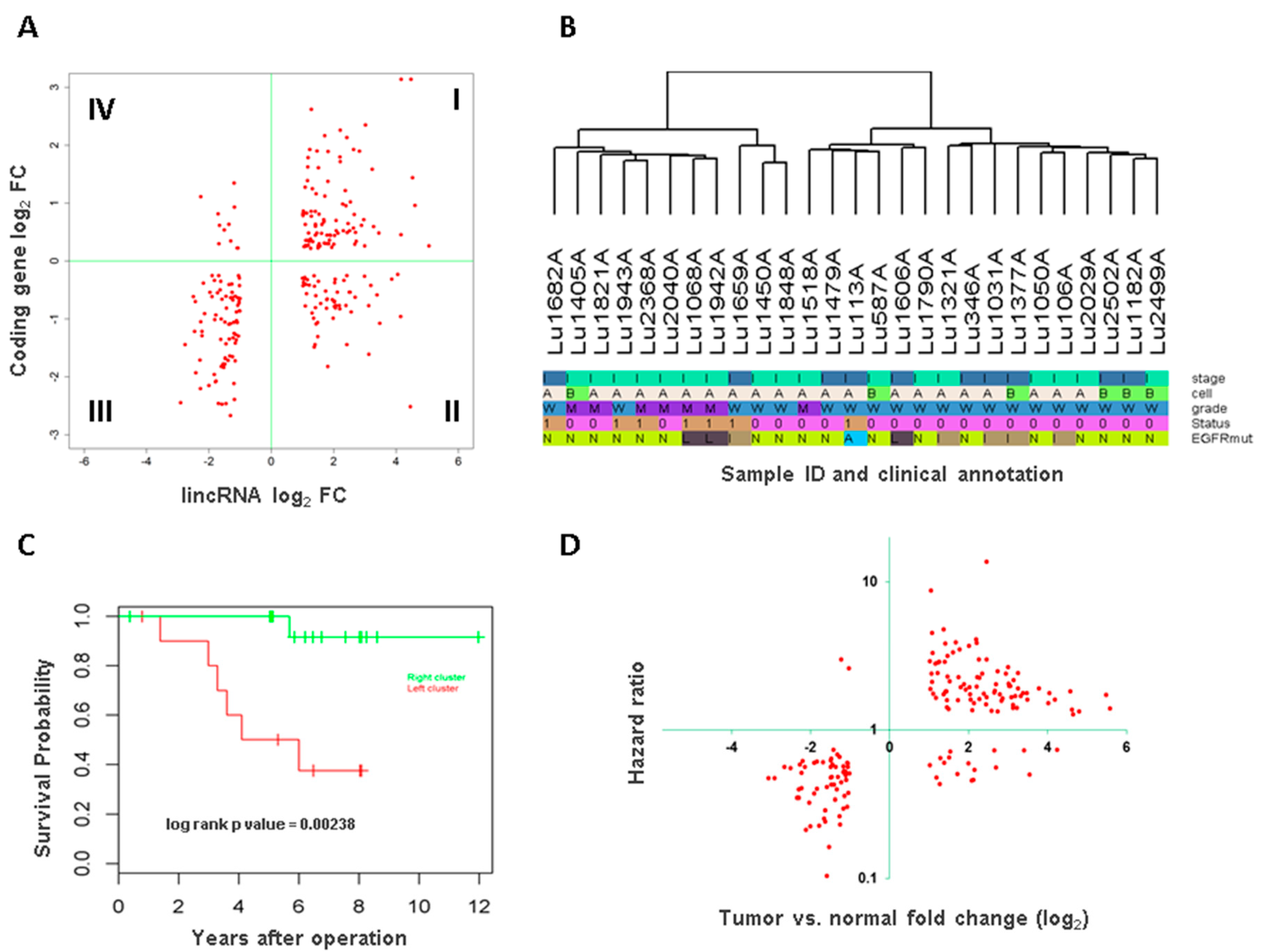
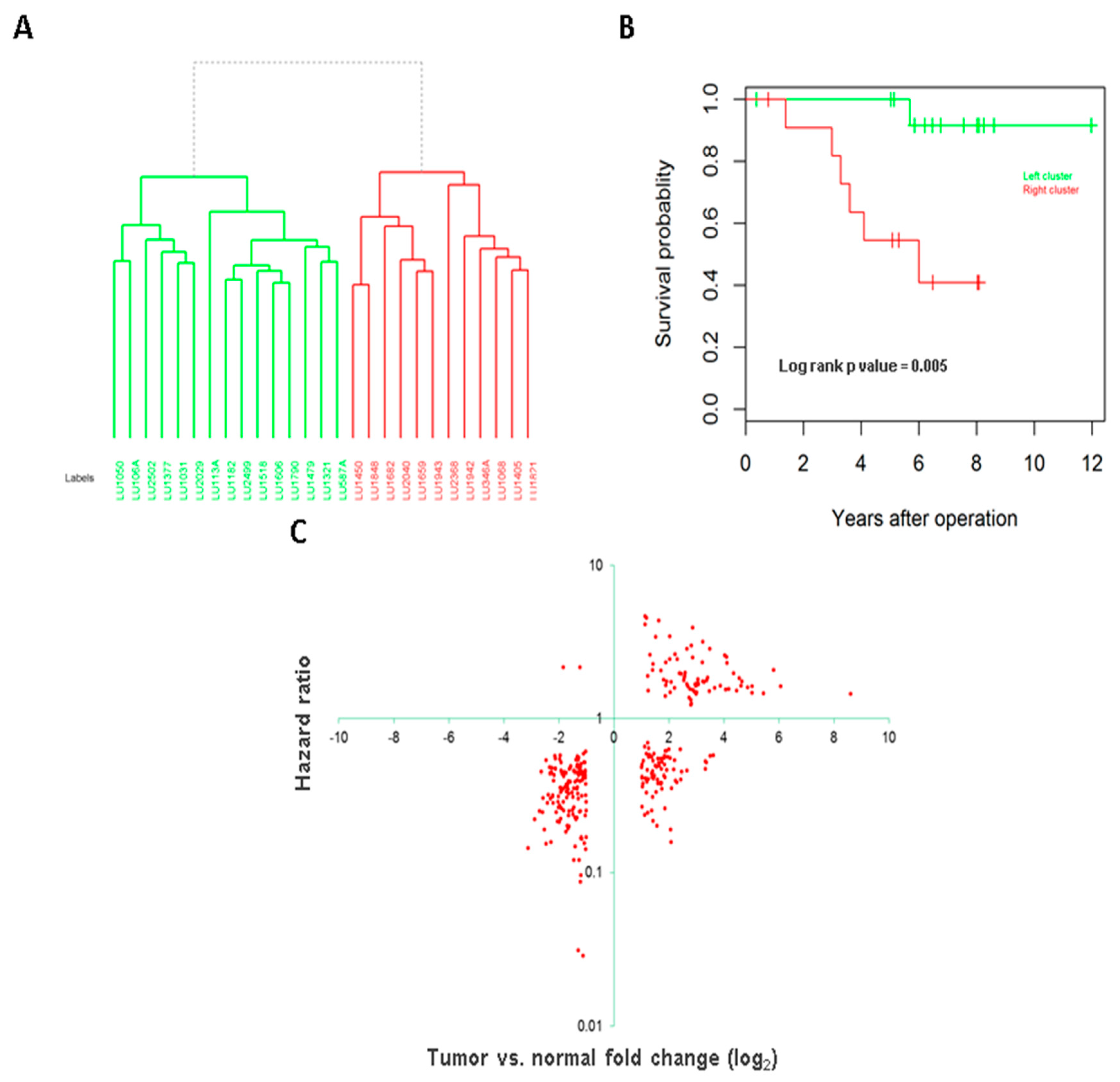
3.3. Association of Long Intergenic Non-Coding RNA with Tumor Aggressiveness and Patient Survival
3.4. Long Intergenic Non-Coding RNAs Associated with Both Cancer Development and Patient Survival
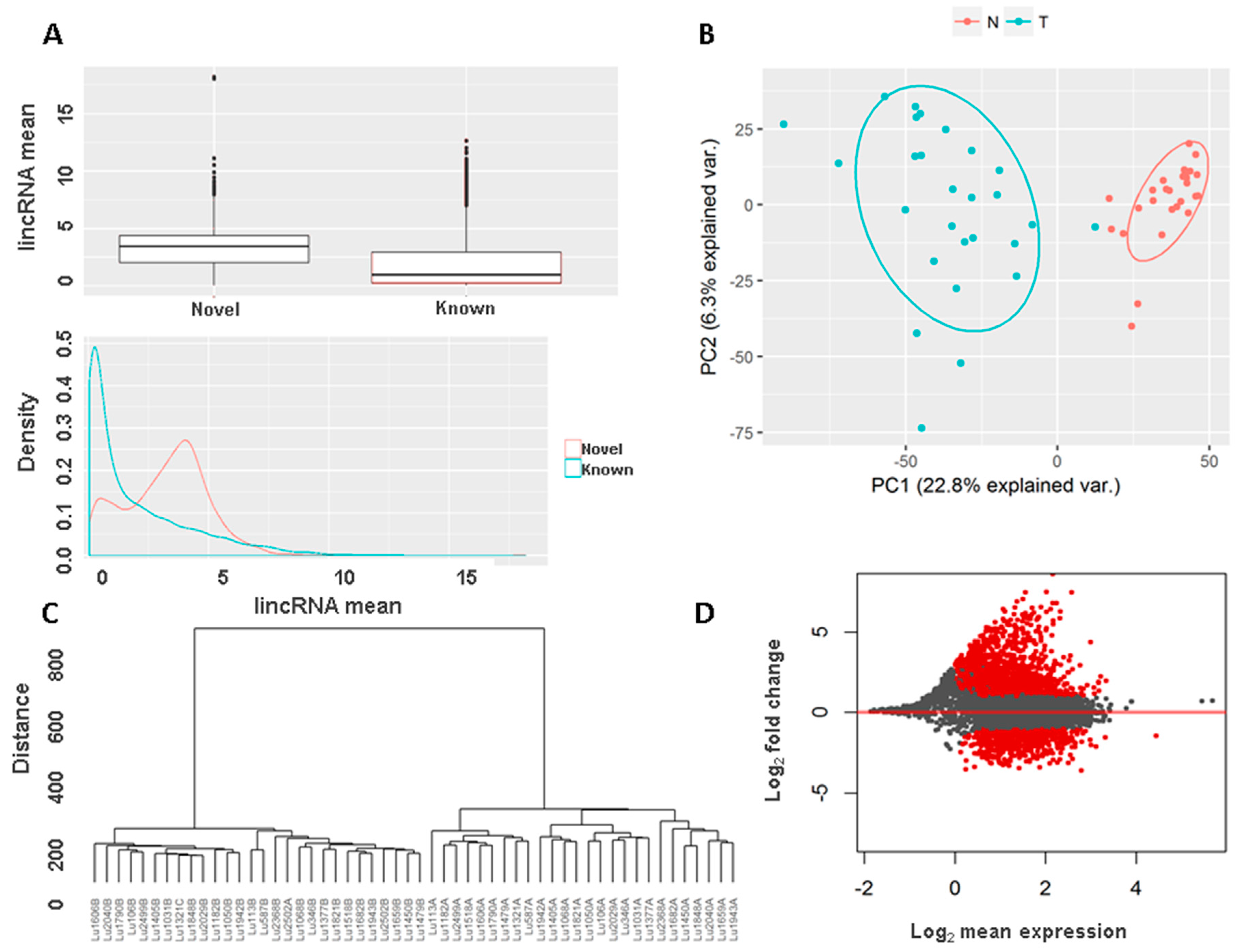
3.5. Novel Long Intergenic Non-Coding RNAs and Their Clinical Associations
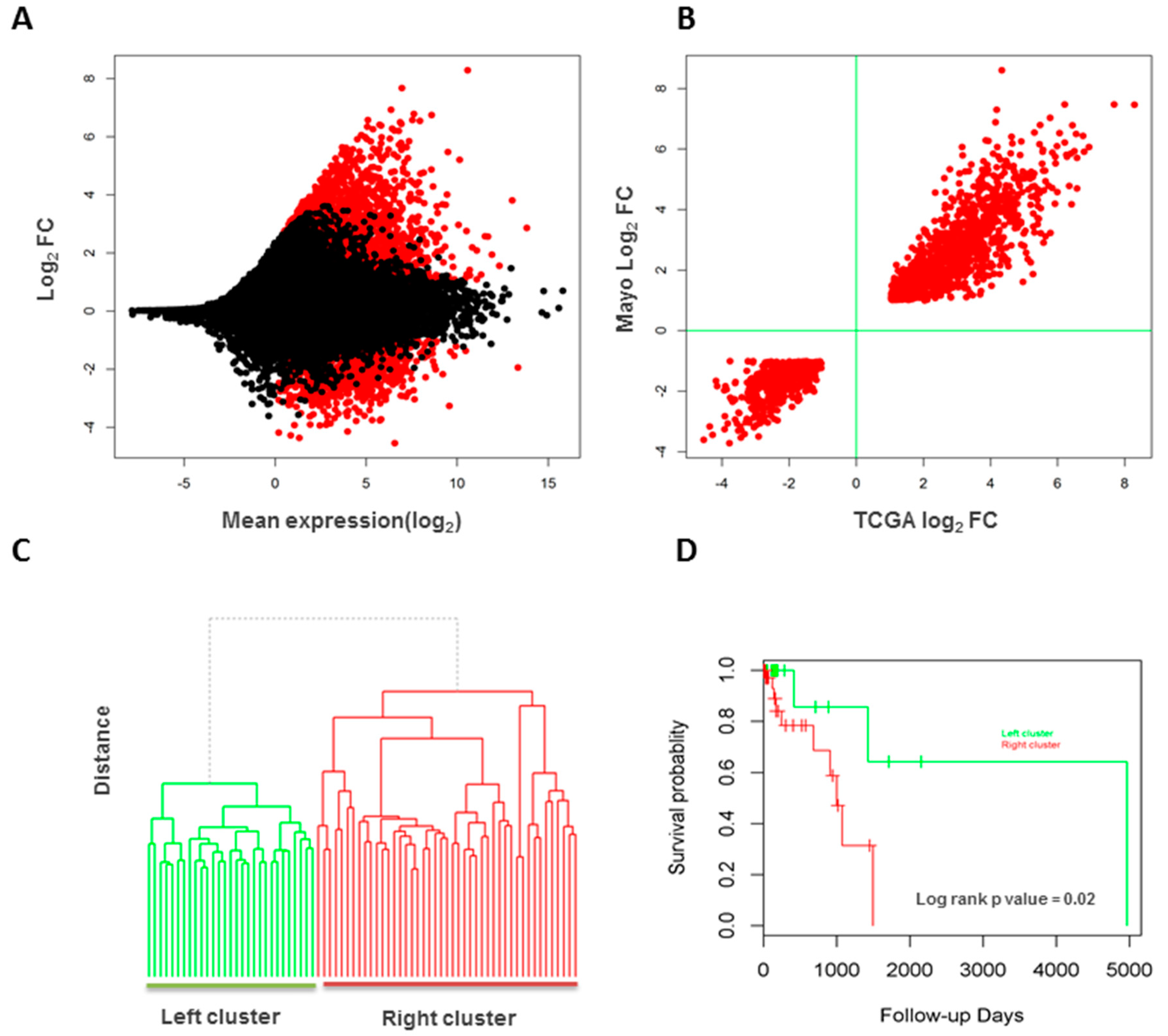
3.6. Validation of Long Intergenic Non-Coding RNAs in the Cancer Genome Atlas Data
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, P.N.; Forey, B.A.; Coombs, K.J.; Lipowicz, P.J.; Appleton, S. Time trends in never smokers in the relative frequency of the different histological types of lung cancer, in particular adenocarcinoma. Regul. Toxicol. Pharmacol. 2016, 74, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Pallis, A.G.; Syrigos, K.N. Lung cancer in never smokers: Disease characteristics and risk factors. Crit. Rev. Oncol. Hematol. 2013, 88, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Rivera, G.A.; Wakelee, H. Lung Cancer in Never Smokers. Adv. Exp. Med. Biol. 2016, 893, 43–57. [Google Scholar] [PubMed]
- Sun, S.; Schiller, J.H.; Gazdar, A.F. Lung cancer in never smokers—A different disease. Nat. Rev. Cancer 2007, 7, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z. High-throughput long noncoding RNA profiling for diagnostic and prognostic markers in cancer: Opportunities and challenges. Epigenomics 2015, 7, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Yuan, S.; Sun, Z.; Li, Y. Long noncoding and circular RNAs in lung cancer: Advances and perspectives. Epigenomics 2016, 8, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Diederichs, S. Long Noncoding RNAs in Lung Cancer. Curr. Top. Microbiol. Immunol. 2016, 394, 57–110. [Google Scholar] [PubMed]
- White, N.M.; Cabanski, C.R.; Silva-Fisher, J.M.; Dang, H.X.; Govindan, R.; Maher, C.A. Transcriptome sequencing reveals altered long intergenic non-coding RNAs in lung cancer. Genome Biol. 2014, 15, 429. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, L.; Eckloff, B.W.; Deng, B.; Wang, Y.; Wampfler, J.A.; Jang, J.; Wieben, E.D.; Jen, J.; You, M.; et al. Conserved recurrent gene mutations correlate with pathway deregulation and clinical outcomes of lung adenocarcinoma in never-smokers. BMC Med. Genom. 2014, 7, 486. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Nair, A.; Chen, X.; Prodduturi, N.; Wang, J.; Kocher, J.-P. UClncR: Ultrafast and comprehensive long non-coding RNA detection from RNA-seq. Sci. Rep. 2017, 7, 14196. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Mitra, R.; Zhao, M.M.; Fan, W.; Eischen, C.M.; Yin, F.; Zhao, Z. The potential roles of long noncoding RNAs (lncRNA) in glioblastoma development. Mol. Cancer Ther. 2016, 15, 2977–2986. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, H.; Yang, Y.; Kang, L. Long noncoding RNA urothelial carcinoma associated 1 promotes the proliferation and metastasis of human lung tumor cells by regulating microRNA-144. Oncol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Jin, L.; Zhang, J.; Yin, Y.; Quan, C.; Hu, Y.; Feng, Y.; Liu, H.; Fei, B.; Mao, Y.; et al. LncRNA-UCA1 enhances cell proliferation and 5-fluorouracil resistance in colorectal cancer by inhibiting miR-204-5p. Sci. Rep. 2016, 6, 23892. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhou, N.; Watabe, K.; Lu, Z.; Wu, F.; Xu, M.; Mo, Y.Y. Long non-coding RNA UCA1 promotes breast tumor growth by suppression of p27 (Kip1). Cell Death Dis. 2014, 5, e1008. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, W.; Yang, C.; Wu, W.; Wu, S.; Qin, X.; Li, X. Long non-coding RNA UCA1a(CUDR) promotes proliferation and tumorigenesis of bladder cancer. Int. J. Oncol. 2012, 41, 276–284. [Google Scholar] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Loewen, G.; Jayawickramarajah, J.; Zhuo, Y.; Shan, B. Functions of lncRNA HOTAIR in lung cancer. J. Hematol. Oncol. 2014, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Xu, G. Clinical value of lncRNA MALAT1 as a prognostic marker in human cancer: Systematic review and meta-analysis. BMJ Open 2015, 5, e008653. [Google Scholar] [CrossRef] [PubMed]
- Beane, J.; Vick, J.; Schembri, F.; Anderlind, C.; Gower, A.; Campbell, J.; Luo, L.; Zhang, X.H.; Xiao, J.; Alekseyev, Y.O.; et al. Characterizing the impact of smoking and lung cancer on the airway transcriptome using RNA-Seq. Cancer Prev. Res. 2011, 4, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Nogueira Jorge, N.A.; Wajnberg, G.; Ferreira, C.G.; de Sa Carvalho, B.; Passetti, F. snoRNA and piRNA expression levels modified by tobacco use in women with lung adenocarcinoma. PLoS ONE 2017, 12, e0183410. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bi, L.; Shi, Z.; Sun, Y.; Lin, Y.; Shao, H.; Zhu, Z. RNA-Seq analysis of non-small cell lung cancer in female never-smokers reveals candidate cancer-associated long non-coding RNAs. Pathol. Res. Pract. 2016, 212, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Amstislavskiy, V.; Risch, T.; Schuette, M.; Dokel, S.; Ralser, M.; Balzereit, D.; Lehrach, H.; Yaspo, M.L. Influence of RNA extraction methods and library selection schemes on RNA-seq data. BMC Genom. 2014, 15, 675. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; He, X.; Hoadley, K.A.; Parker, J.S.; Hayes, D.N.; Perou, C.M. Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genom. 2014, 15, 419. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wang, Z.; Nair, A.; Song, W.; Yang, P.; Zhang, X.; Sun, Z. Comprehensive Profiling of lincRNAs in Lung Adenocarcinoma of Never Smokers Reveals Their Roles in Cancer Development and Prognosis. Genes 2017, 8, 321. https://doi.org/10.3390/genes8110321
Li Y, Wang Z, Nair A, Song W, Yang P, Zhang X, Sun Z. Comprehensive Profiling of lincRNAs in Lung Adenocarcinoma of Never Smokers Reveals Their Roles in Cancer Development and Prognosis. Genes. 2017; 8(11):321. https://doi.org/10.3390/genes8110321
Chicago/Turabian StyleLi, Ying, Zheng Wang, Asha Nair, Wei Song, Ping Yang, Xiaoju Zhang, and Zhifu Sun. 2017. "Comprehensive Profiling of lincRNAs in Lung Adenocarcinoma of Never Smokers Reveals Their Roles in Cancer Development and Prognosis" Genes 8, no. 11: 321. https://doi.org/10.3390/genes8110321
APA StyleLi, Y., Wang, Z., Nair, A., Song, W., Yang, P., Zhang, X., & Sun, Z. (2017). Comprehensive Profiling of lincRNAs in Lung Adenocarcinoma of Never Smokers Reveals Their Roles in Cancer Development and Prognosis. Genes, 8(11), 321. https://doi.org/10.3390/genes8110321