
An Expanded Multi-Organ Disease Phenotype Associated with Mutations in YARS

Abstract
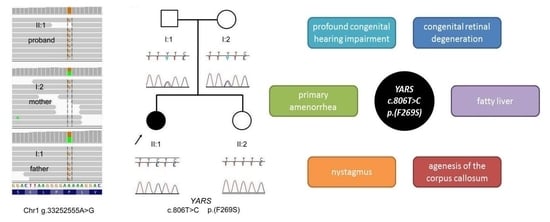
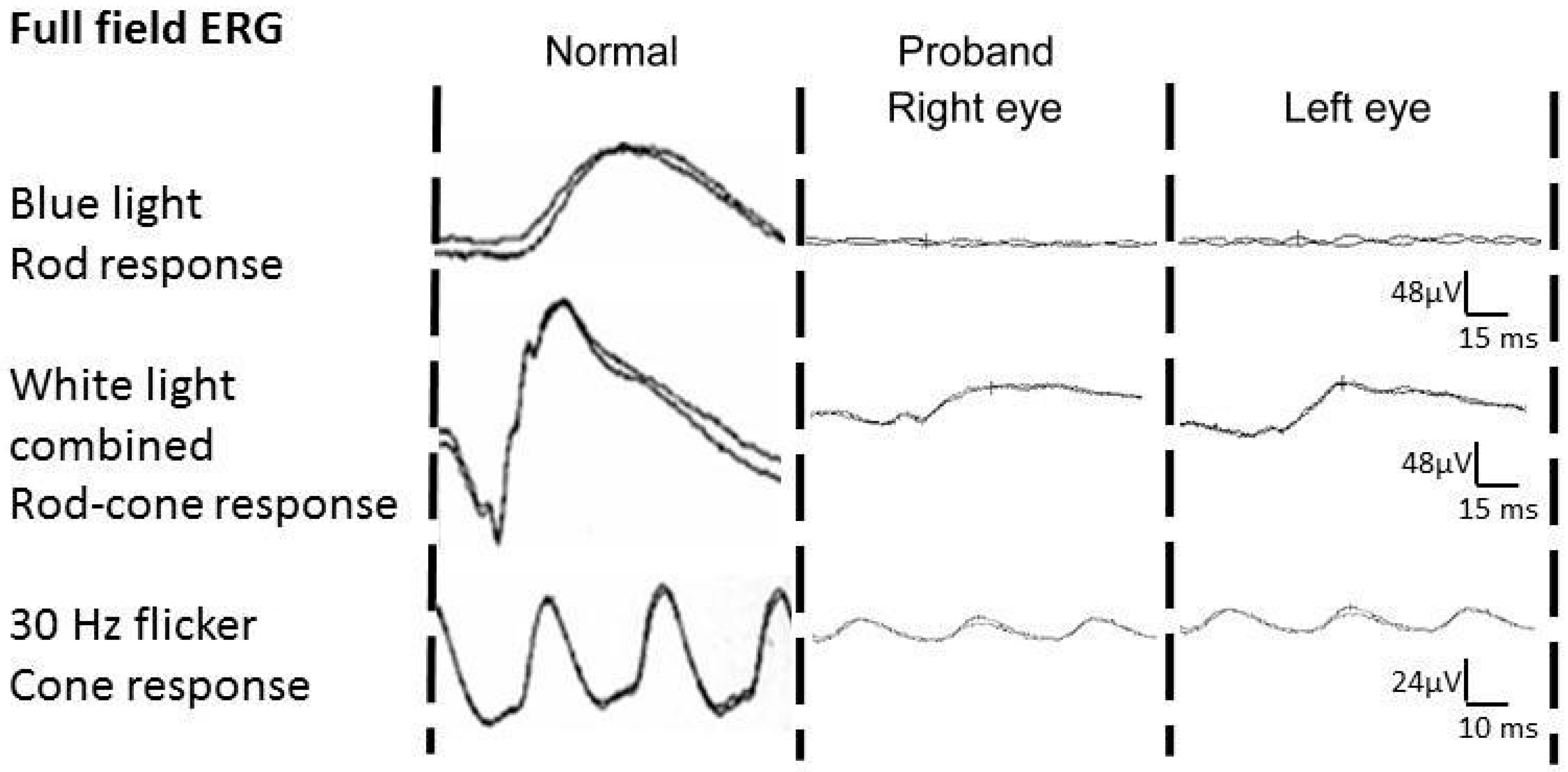
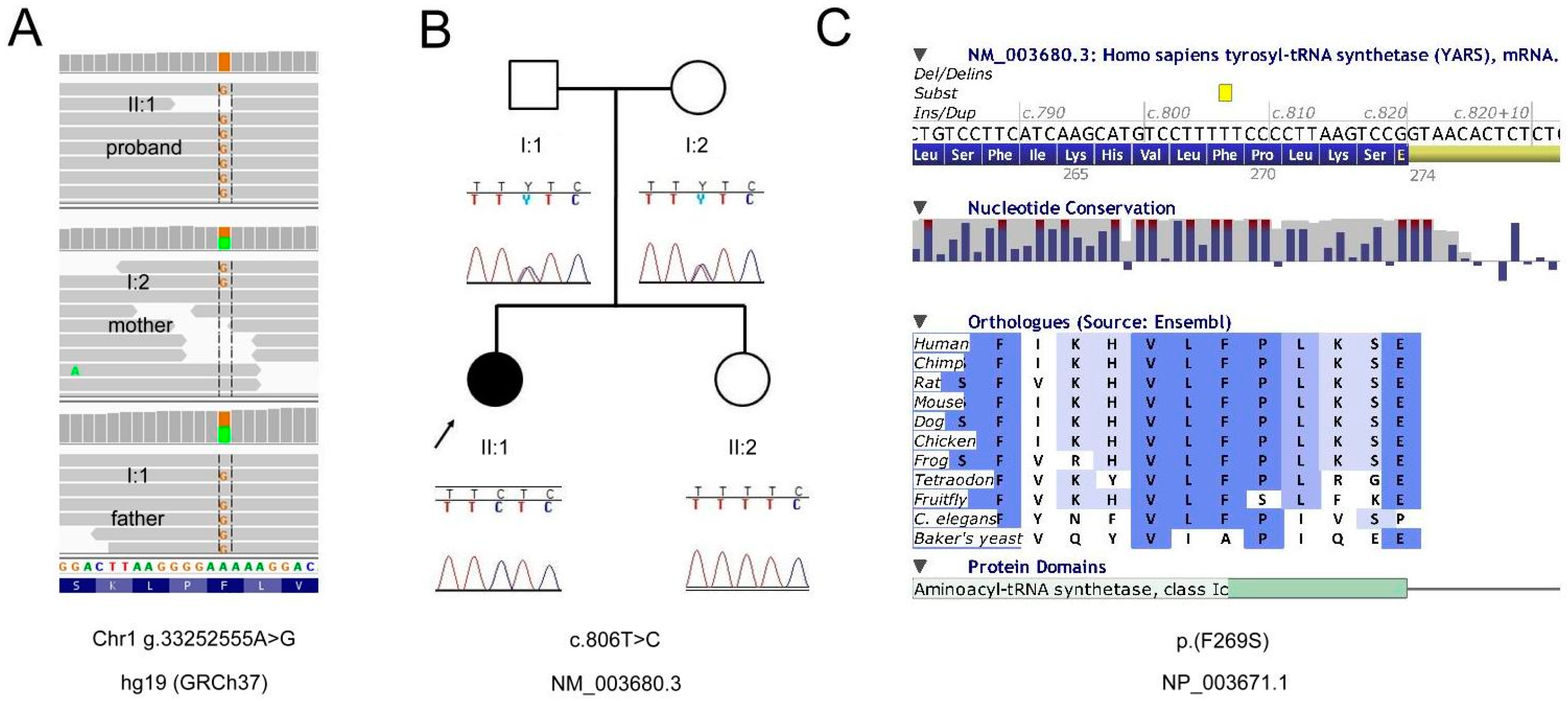
:
1. Introduction
Clinical Case Report
2. Materials and Methods
2.1. Genetic Analyses
2.2. Exome Report
3. Results and Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berg, J.M. Biochemistry, revised edition. Chem. Eng. News 2001, 79, 130. [Google Scholar]
- Park, S.G.; Schimmel, P.; Kim, S. Aminoacyl tRNA synthetases and their connections to disease. Proc. Natl Acad. Sci. USA 2008, 105, 11043–11049. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Schuman, R.; Antonellis, A. Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum. Mol. Genet. 2017, 26, R114–R127. [Google Scholar] [CrossRef] [PubMed]
- Jordanova, A.; Irobi, J.; Thomas, F.P.; Van Dijck, P.; Meerschaert, K.; Dewil, M.; Dierick, I.; Jacobs, A.; De Vriendt, E.; Guergueltcheva, V.; et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006, 38, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Nowaczyk, M.J.; Huang, L.; Tarnopolsky, M.; Schwartzentruber, J.; Majewski, J.; Bulman, D.E.; Hartley, T.; Boycott, K.M. A novel multisystem disease associated with recessive mutations in the tyrosyl-tRNA synthetase (YARS) gene. Am. J. Med. Genet. Part A 2017, 173, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, C.G.; Cox, J.; Springell, K.; Hampshire, D.J.; Mohamed, M.D.; McKibbin, M.; Stern, R.; Raymond, F.L.; Sandford, R.; Malik Sharif, S.; et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet. 2006, 78, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.D.; MacDonald, R.J. Evolutionary silencing of the human elastase I gene (ELA1). Hum. Mol. Genet. 1997, 6, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Talas, U.; Dunlop, J.; Khalaf, S.; Leigh, I.M.; Kelsell, D.P. Human elastase 1: Evidence for expression in the skin and the identification of a frequent frameshift polymorphism. J. Investig. Dermatol. 2000, 114, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Peragallo, J.H.; Keller, S.; van der Knaap, M.S.; Soares, B.P.; Shankar, S.P. Retinopathy and optic atrophy: Expanding the phenotypic spectrum of pathogenic variants in the AARS2 gene. Ophthalmic Genet. 2017, 19, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Puffenberger, E.G.; Jinks, R.N.; Sougnez, C.; Cibulskis, K.; Willert, R.A.; Achilly, N.P.; Cassidy, R.P.; Fiorentini, C.J.; Heiken, K.F.; Lawrence, J.J.; et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE 2012, 7, e28936. [Google Scholar] [CrossRef] [PubMed]
- Elo, J.M.; Yadavalli, S.S.; Euro, L.; Isohanni, P.; Gotz, A.; Carroll, C.J.; Valanne, L.; Alkuraya, F.S.; Uusimaa, J.; Paetau, A.; et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum. Mol. Genet. 2012, 21, 4521–4529. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Chisholm, K.M.; Lynch, E.D.; Lee, M.K.; Walsh, T.; Opitz, J.M.; Li, W.; Klevit, R.E.; King, M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 6543–6548. [Google Scholar] [CrossRef] [PubMed]
- Steenweg, M.E.; Ghezzi, D.; Haack, T.; Abbink, T.E.; Martinelli, D.; van Berkel, C.G.; Bley, A.; Diogo, L.; Grillo, E.; Te Water Naude, J.; et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain J. Neurol. 2012, 135, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Belostotsky, R.; Ben-Shalom, E.; Rinat, C.; Becker-Cohen, R.; Feinstein, S.; Zeligson, S.; Segel, R.; Elpeleg, O.; Nassar, S.; Frishberg, Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 2011, 88, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.; Fox, P.L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 2013, 5, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Simons, C.; Griffin, L.B.; Helman, G.; Golas, G.; Pizzino, A.; Bloom, M.; Murphy, J.L.; Crawford, J.; Evans, S.H.; Topper, S.; et al. Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am. J. Hum. Genet. 2015, 96, 675–681. [Google Scholar] [CrossRef] [PubMed]
- McMillan, H.J.; Schwartzentruber, J.; Smith, A.; Lee, S.; Chakraborty, P.; Bulman, D.E.; Beaulieu, C.L.; Majewski, J.; Boycott, K.M.; Geraghty, M.T. Compound heterozygous mutations in glycyl-tRNA synthetase are a proposed cause of systemic mitochondrial disease. BMC Med. Genet. 2014, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Van Meel, E.; Wegner, D.J.; Cliften, P.; Willing, M.C.; White, F.V.; Kornfeld, S.; Cole, F.S. Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med. Genet. 2013, 14, 106. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
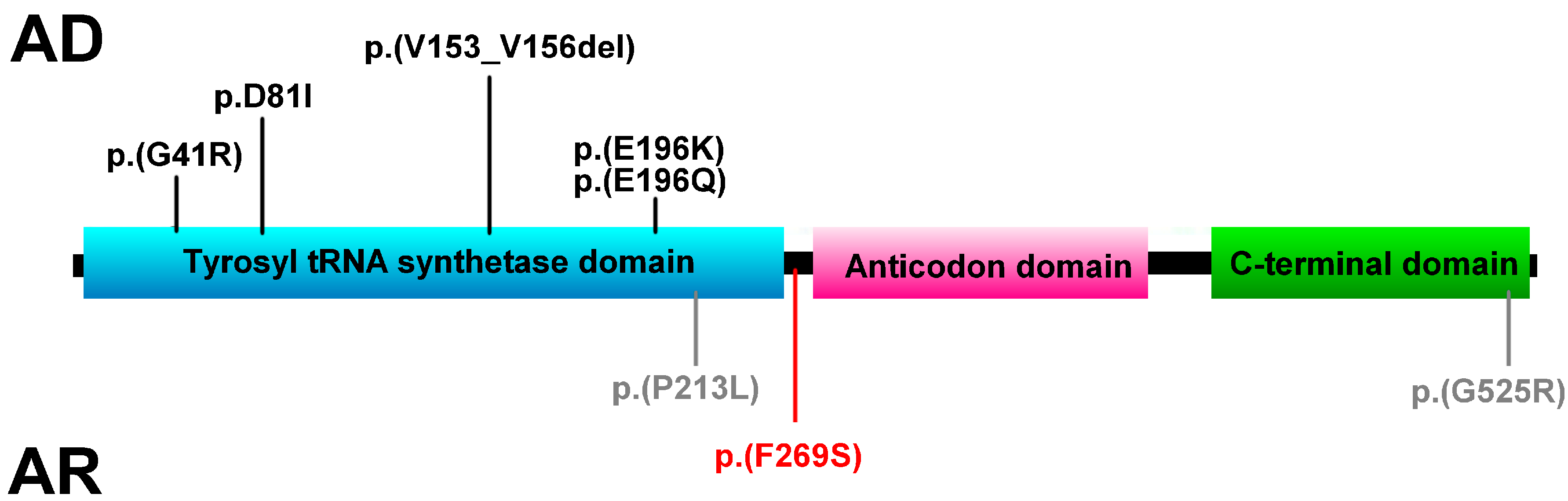{kind=link}
| This Study | Nowaczyk et al. | DI-CMT Syndrome | Perrault Syndrome 2 | ||
|---|---|---|---|---|---|
| Patient no. | II:1 | Sibling 1 | Sibling 2 | Multiple patients | Multiple patients |
| Gender | F | M | F | F/M | F |
| Mutations | YARS p.(F269S)/p.(F269S) | YARS p.(P213L)/p.(G525R) | YARS p.(P213L)/p.(G525R) | YARS p.(G41R)/+ p.(E196K)/+ p.(D81I)/+ p.(V153_V156del)/+ | HARS2 (Many) |
| Abnormality of corpus callosum | +++ (Agenesis) | + (Thinning) | - | - (Thinning observed in other forms of CMT) | - |
| Fatty liver | + (Transient) | ++ | ++ | - | - |
| Specific facial features | - | + | + | - | - |
| Hypotonia | + | + | + | + | - |
| Retnititis pigmentosa | + | - | - | - | + |
| Deafness | + | - | - | - | + |
| Primary amenorrhea | + | n.a. | n.d. | - | + |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tracewska-Siemiątkowska, A.; Haer-Wigman, L.; Bosch, D.G.M.; Nickerson, D.; Bamshad, M.J.; University of Washington Center for Mendelian Genomics; Van de Vorst, M.; Rendtorff, N.D.; Möller, C.; Kjellström, U.; et al. An Expanded Multi-Organ Disease Phenotype Associated with Mutations in YARS. Genes 2017, 8, 381. https://doi.org/10.3390/genes8120381
Tracewska-Siemiątkowska A, Haer-Wigman L, Bosch DGM, Nickerson D, Bamshad MJ, University of Washington Center for Mendelian Genomics, Van de Vorst M, Rendtorff ND, Möller C, Kjellström U, et al. An Expanded Multi-Organ Disease Phenotype Associated with Mutations in YARS. Genes. 2017; 8(12):381. https://doi.org/10.3390/genes8120381
Chicago/Turabian StyleTracewska-Siemiątkowska, Anna, Lonneke Haer-Wigman, Danielle G. M. Bosch, Deborah Nickerson, Michael J. Bamshad, University of Washington Center for Mendelian Genomics, Maartje Van de Vorst, Nanna Dahl Rendtorff, Claes Möller, Ulrika Kjellström, and et al. 2017. "An Expanded Multi-Organ Disease Phenotype Associated with Mutations in YARS" Genes 8, no. 12: 381. https://doi.org/10.3390/genes8120381
APA StyleTracewska-Siemiątkowska, A., Haer-Wigman, L., Bosch, D. G. M., Nickerson, D., Bamshad, M. J., University of Washington Center for Mendelian Genomics, Van de Vorst, M., Rendtorff, N. D., Möller, C., Kjellström, U., Andréasson, S., Cremers, F. P. M., & Tranebjærg, L. (2017). An Expanded Multi-Organ Disease Phenotype Associated with Mutations in YARS. Genes, 8(12), 381. https://doi.org/10.3390/genes8120381