Identification of Inherited Retinal Disease-Associated Genetic Variants in 11 Candidate Genes

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subject and Clinical Evaluation
2.2. Genetic Analysis
2.3. Variant Prioritization
2.4. Data Sharing
3. Results
3.1. Score 4 Candidate IRD Gene
3.2. Score 3 Candidate IRD Genes
3.3. Score 2 Candidate IRD Genes
3.4. Score 1 Candidate Gene
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Francis, P.J. Genetics of inherited retinal disease. J. R. Soc. Med. 2006, 99, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Finger, R.P.; Fimmers, R.; Holz, F.G.; Scholl, H.P. Prevalence and causes of registered blindness in the largest federal state of Germany. Br. J. Ophthalmol. 2011, 95, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef] [PubMed]
- Koenekoop, R.K.; Sui, R.; Sallum, J.; van den Born, L.I.; Ajlan, R.; Khan, A.; den Hollander, A.I.; Cremers, F.P.; Mendola, J.D.; Bittner, A.K.; et al. Oral 9-cis retinoid for childhood blindness due to leber congenital amaurosis caused by RPE65 or LRAT mutations: An open-label phase 1b trial. Lancet 2014, 384, 1513–1520. [Google Scholar] [CrossRef]
- MacLaren, R.E.; Groppe, M.; Barnard, A.R.; Cottriall, C.L.; Tolmachova, T.; Seymour, L.; Clark, K.R.; During, M.J.; Cremers, F.P.; Black, G.C.; et al. Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet 2014, 383, 1129–1137. [Google Scholar] [CrossRef]
- Bennett, J.; Wellman, J.; Marshall, K.A.; McCague, S.; Ashtari, M.; DiStefano-Pappas, J.; Elci, O.U.; Chung, D.C.; Sun, J.; Wright, J.F.; et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: A follow-on phase 1 trial. Lancet 2016, 388, 661–672. [Google Scholar] [CrossRef]
- RetNet. Available online: http://www.sph.uth.tmc.edu/RetNet/ (accessed on 1 September 2017).
- Patel, N.; Aldahmesh, M.A.; Alkuraya, H.; Anazi, S.; Alsharif, H.; Khan, A.O.; Sunker, A.; Al-Mohsen, S.; Abboud, E.B.; Nowilaty, S.R.; et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Beryozkin, A.; Shevah, E.; Kimchi, A.; Mizrahi-Meissonnier, L.; Khateb, S.; Ratnapriya, R.; Lazar, C.H.; Blumenfeld, A.; Ben-Yosef, T.; Hemo, Y.; et al. Whole exome sequencing reveals mutations in known retinal disease genes in 33 out of 68 israeli families with inherited retinopathies. Sci. Rep. 2015, 5, 13187. [Google Scholar] [CrossRef] [PubMed]
- Weisschuh, N.; Mayer, A.K.; Strom, T.M.; Kohl, S.; Glockle, N.; Schubach, M.; Andreasson, S.; Bernd, A.; Birch, D.G.; Hamel, C.P.; et al. Mutation detection in patients with retinal dystrophies using targeted next generation sequencing. PLoS ONE 2016, 11, e0145951. [Google Scholar] [CrossRef] [PubMed]
- Haer-Wigman, L.; van Zelst-Stams, W.A.G.; Pfund, R.; van den Born, L.I.; Klaver, C.C.W.; Verheij, J.B.G.M.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.F.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef] [PubMed]
- European Retinal Disease Consortium. Available online: www.ERDC.info (accessed on 1 September 2017).
- Estrada-Cuzcano, A.; Neveling, K.; Kohl, S.; Banin, E.; Rotenstreich, Y.; Sharon, D.; Falik-Zaccai, T.C.; Hipp, S.; Roepman, R.; Wissinger, B.; et al. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am. J. Hum. Genet. 2012, 90, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Ozgul, R.K.; Siemiatkowska, A.M.; Yucel, D.; Myers, C.A.; Collin, R.W.; Zonneveld, M.N.; Beryozkin, A.; Banin, E.; Hoyng, C.B.; van den Born, L.I.; et al. Exome sequencing and cis-regulatory mapping identify mutations in MAK, a gene encoding a regulator of ciliary length, as a cause of retinitis pigmentosa. Am. J. Hum. Genet. 2011, 89, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Peluso, I.; Conte, I.; Testa, F.; Dharmalingam, G.; Pizzo, M.; Collin, R.W.; Meola, N.; Barbato, S.; Mutarelli, M.; Ziviello, C.; et al. The ADAMTS18 gene is responsible for autosomal recessive early onset severe retinal dystrophy. Orphanet J. Rare Dis. 2013, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosing, S.; Rohrschneider, K.; Beryozkin, A.; Sharon, D.; Weisschuh, N.; Staller, J.; Kohl, S.; Zelinger, L.; Peters, T.A.; Neveling, K.; et al. Mutations in RAB28, encoding a farnesylated small GTPase, are associated with autosomal-recessive cone-rod dystrophy. Am. J. Hum. Genet. 2013, 93, 110–117. [Google Scholar] [CrossRef] [PubMed]
- AgileMultildeogram. Available online: http://dna.leeds.ac.uk/agile/AgileMultiIdeogram/ (accessed on 1 September 2017).
- Thermo Fisher. Available online: https://www.thermofisher.com/us/en/home/life-science/microarray-analysis/microarray-analysis-instruments-software-services/microarray-analysis-software/chromosome-analysis-suite.html (accessed on 1 September 2017).
- Homozygosity Mapper. Available online: http://www.homozygositymapper.org/ (accessed on 1 September 2017).
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 15, 860–921. [Google Scholar] [CrossRef] [PubMed]
- PLINK. Available online: http://zzz.bwh.harvard.edu/plink/ibdibs.shtml (accessed on 1 September 2017).
- Exome Aggregation Consortium. Available online: http://exac.broadinstitute.org/ (accessed on 1 September 2017).
- Yet Another Scientific Artificial Reality Application. Available online: http://www.yasara.org/ (accessed on 1 September 2017).
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Primer3. Available online: http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi (accessed on 1 September 2017).
- STRING. Available online: https://string-db.org/ (accessed on 1 September 2017).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Rolland, T.; Taşan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Kanno, E.; Saegusa, C.; Ogata, Y.; Kuroda, T.S. Slp4-a/granuphilin-a regulates dense-core vesicle exocytosis in PC12 cells. J. Biol. Chem. 2001, 277, 39673–39678. [Google Scholar] [CrossRef] [PubMed]
- Fuerst, P.G.; Bruce, F.; Tian, M.; Wei, W.; Elstrott, J.; Feller, M.B.; Erskine, L.; Singer, J.H.; Burgess, R.W. DSCAM and DSCAML1 function in self-avoidance in multiple cell types in the developing mouse retina. Neuron 2009, 64, 484–497. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, G.B.; Long, S.S.; Fleming, H.; Li, W.; Fuerst, P.G. DSCAM localization and function at the mouse cone synapse. J. Comp. Neurol. 2014, 522, 2609–2633. [Google Scholar] [CrossRef] [PubMed]
- Yariz, K.O.; Duman, D.; Zazo Seco, C.; Dallman, J.; Huang, M.; Peters, T.A.; Sirmaci, A.; Lu, N.; Schraders, M.; Skromne, I.; et al. Mutations in OTOGL, encoding the inner ear protein otogelin-like, cause moderate sensorineural hearing loss. Am. J. Hum. Genet. 2012, 91, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Louha, M.; Loundon, N.; Michalski, N.; Verpy, E.; Smagghe, L.; Hardelin, J.P.; Rouillon, I.; Jonard, L.; Couderc, R.; et al. Biallelic nonsense mutations in the otogelin-like gene (OTOGL) in a child affected by mild to moderate hearing impairment. Gene 2013, 527, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Roelfsema, J.H.; White, S.J.; Ariyurek, Y.; Bartholdi, D.; Niedrist, D.; Papadia, F.; Bacino, C.A.; den Dunnen, J.T.; van Ommen, G.J.; Breuning, M.H.; et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: Mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 2005, 76, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Hennig, A.K.; Peng, G.H.; Chen, S. Transcription coactivators p300 and CBP are necessary for photoreceptor-specific chromatin organization and gene expression. PLoS ONE 2013, 8, e69721. [Google Scholar] [CrossRef] [PubMed]
- Kawase, R.; Nishimura, Y.; Ashikawa, Y.; Sasagawa, S.; Murakami, S.; Yuge, M.; Okabe, S.; Kawaguchi, K.; Yamamoto, H.; Moriyuki, K.; et al. EP300 protects from light-induced retinopathy in Zebrafish. Front. Pharmacol. 2016, 7, 126. [Google Scholar] [CrossRef] [PubMed]
- Roosing, S.; Collin, R.W.; den Hollander, A.I.; Cremers, F.P.; Siemiatkowska, A.M. Prenylation defects in inherited retinal diseases. J. Med. Genet. 2014, 51, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Siemiatkowska, A.M.; van den Born, L.I.; van Hagen, P.M.; Stoffels, M.; Neveling, K.; Henkes, A.; Kipping-Geertsema, M.; Hoefsloot, L.H.; Hoyng, C.B.; Simon, A.; et al. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology 2013, 120, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kokame, K.; Okuda, T.; Nakajo, Y.; Yanamoto, H.; Miyata, T. NDRG4 protein-deficient mice exhibit spatial learning deficits and vulnerabilities to cerebral ischemia. J. Biol. Chem. 2011, 286, 26158–26165. [Google Scholar] [CrossRef] [PubMed]
- Mullally, M.; Albrecht, C.; Horton, M.; Laboissonniere, L.A.; Goetz, J.J.; Chowdhury, R.; Manning, A.; Wester, A.K.; Bose, Q.; Trimarchi, J.M. Expression profiling of developing Zebrafish retinal cells. Zebrafish 2016, 13, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Sergouniotis, P.I.; Davidson, A.E.; Mackay, D.S.; Li, Z.; Yang, X.; Plagnol, V.; Moore, A.T.; Webster, A.R. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am. J. Hum. Genet. 2011, 89, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Scheetz, T.E.; Mullins, R.F.; DeLuca, A.P.; Hoffmann, J.M.; Johnston, R.M.; Jacobson, S.G.; Sheffield, V.C.; Stone, E.M. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2011, 108, E569–E576. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; O’Sullivan, J.; Williams, S.G.; Lamb, J.A.; Panda, B.; Sergouniotis, P.I.; Gillespie, R.L.; Daiger, S.P.; et al. Molecular findings from 537 individuals with inherited retinal disease. J. Med. Genet. 2016, 53, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Tatour, Y.; Sanchez-Navarro, I.; Chervinsky, E.; Hakonarson, H.; Gawi, H.; Tahsin-Swafiri, S.; Leibu, R.; Lopez-Molina, M.I.; Fernandez-Sanz, G.; Ayuso, C.; et al. Mutations in SCAPER cause autosomal recessive retinitis pigmentosa with intellectual disability. J. Med. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. Genematcher: A matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Sun, V.; Tuan, H.F.; Keser, V.; Wang, K.; Ren, H.; Lopez, I.; Zaneveld, J.E.; Siddiqui, S.; et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J. Med. Genet. 2013, 50, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Bujakowska, K.M.; Fernandez-Godino, R.; Place, E.; Consugar, M.; Navarro-Gomez, D.; White, J.; Bedoukian, E.C.; Zhu, X.; Xie, H.M.; Gai, X.; et al. Copy-number variation is an important contributor to the genetic causality of inherited retinal degenerations. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Nakaya, N.; Chavali, V.R.; Ma, Z.; Jiao, X.; Sieving, P.A.; Riazuddin, S.; Tomarev, S.I.; Ayyagari, R.; Riazuddin, S.A.; et al. A mutation in ZNF513, a putative regulator of photoreceptor development, causes autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010, 87, 400–409. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
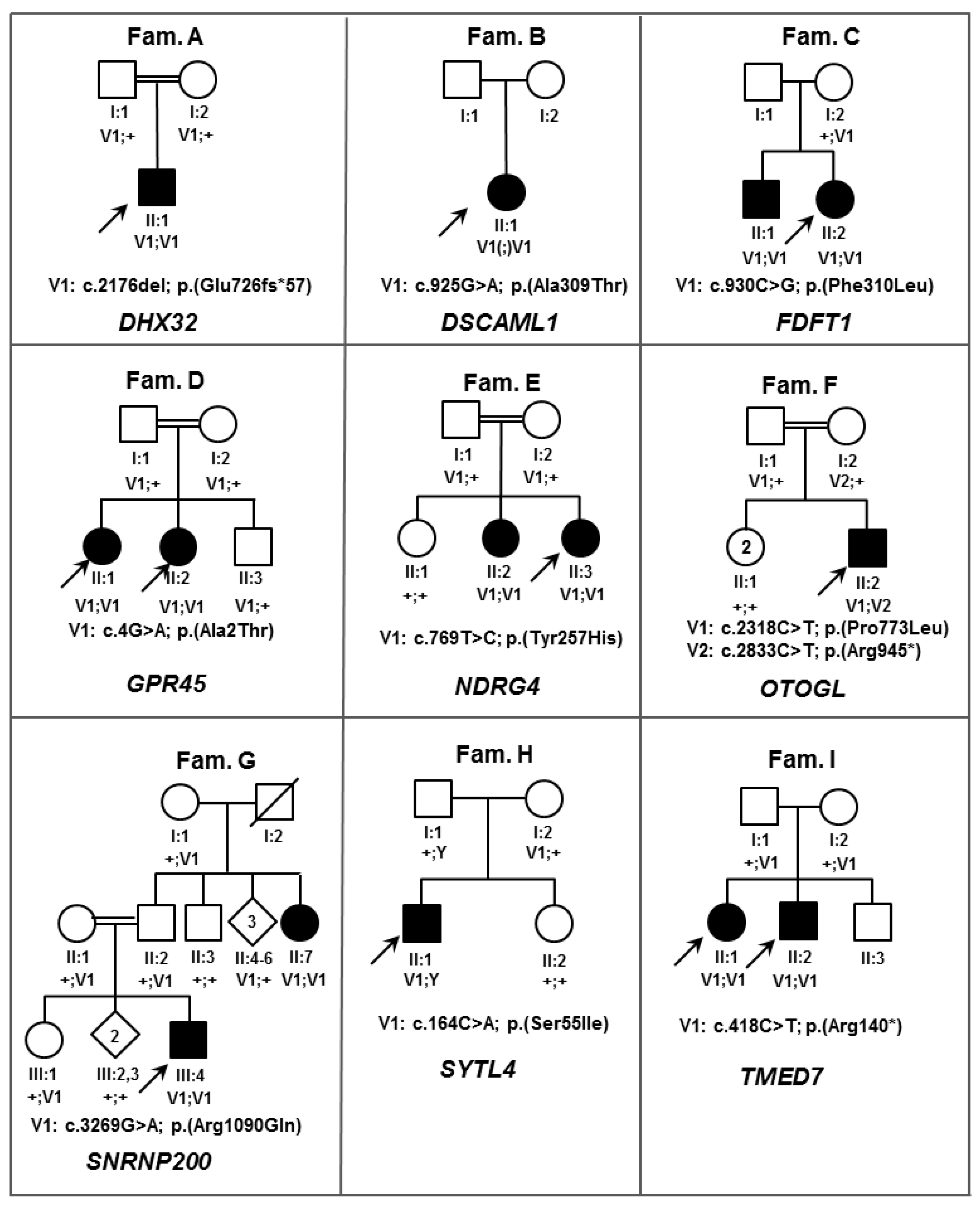
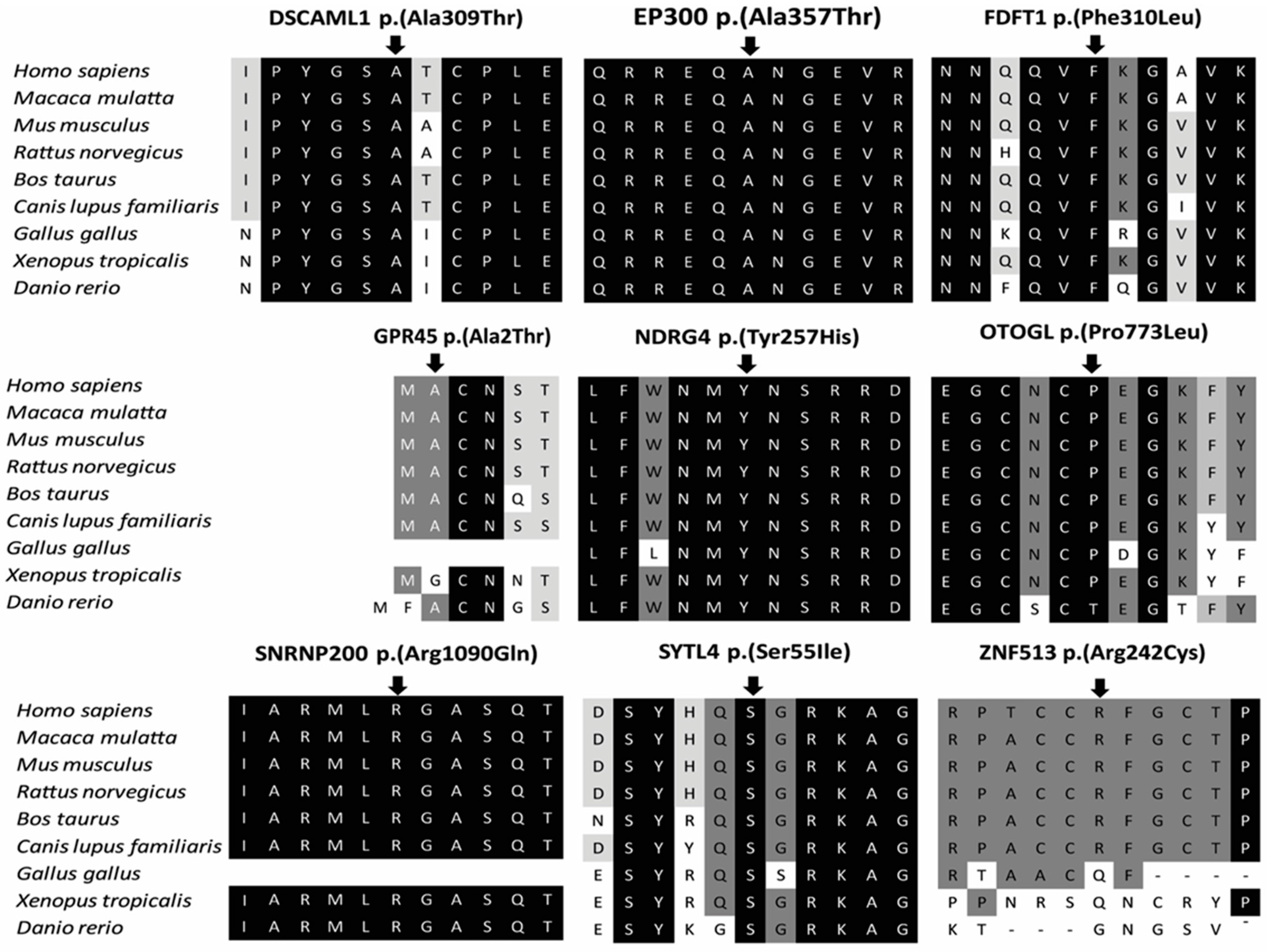
| Gene name | Mode of Inheritance | Zygosity | DNA Variant | Protein Variant | CADD_Phred | ExAC $ AC/AN/HA/AF | Described Protein Interaction | Known IRD-Associated Gene | Score * | Interpretation According to ACMG Guidelines # | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Type of Evidence | Classification | ||||||||||
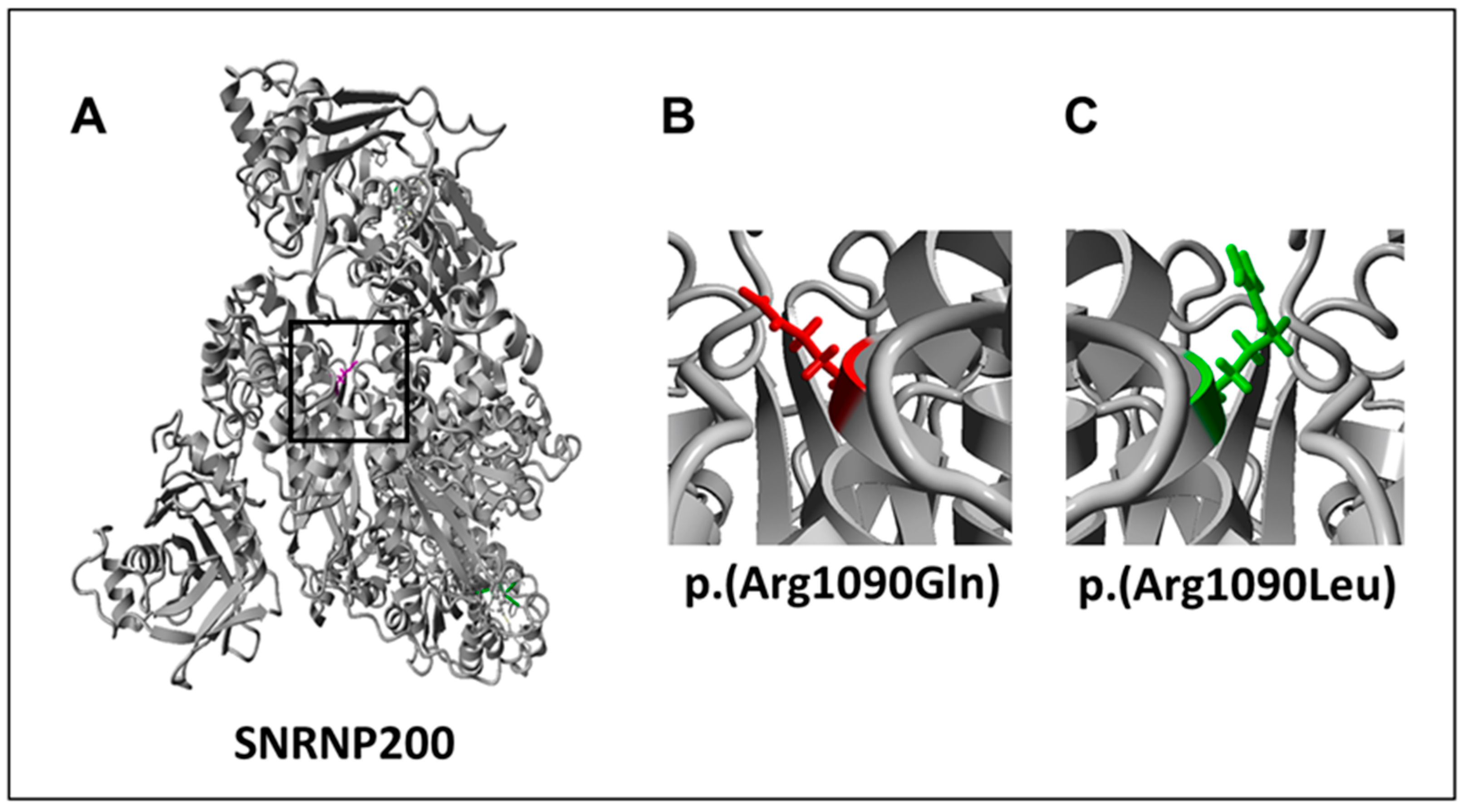
| SNRNP200 | AR | Hom | c.3269G>A | p.(Arg1090Gln) | 29.6 | 3/121,400/0/0.00002471 | PRPF4; PRPF6; PRPF8 [27] | yes | 4 | PM1, PM5, PP1, PP2, PP3, PP4 | LP |
| DHX32 | AR | Hom | c.2176del | p.(Glu726Asnfs*57) | NA | 2/121,276/0/0.00001649 | FAM161A [31] | no | 3 | PVS1, PM4 | P |
| ZNF513 | AD | Het | c.724C>T | p.(Arg242Cys) | 17.61 | 0/121,412/0/0 | OPN1SW; PDE6A [27] | yes | 3 | PP2, PM2 | US |
| DSCAML1 | AR | Hom | c.925G>A | p.(Ala309Thr) | 34 | 2/91,000/0/0.00002198 | no | 2 | PP3 | P | |
| EP300 | AD | Het | c.1069G>A | p.(Ala357Thr) | 36 | 0/121,412/0/0 | no | 2 | PP1, PP3, PM2 | US | |
| FDFT1 | AR | Hom | c.930C>G | p.(Phe310Leu) | 19.38 | 0/121,412/0/0 | no | 2 | PP1, PP3, PM2 | US | |
| NDRG4 | AR | Hom | c.769T>C | p.(Tyr257His) | 27.4 | 0/121,412/0/0 | no | 2 | PP1, PP3, PM2 | US | |
| OTOGL | AR | Het | c.2318C>T | p.(Pro773Leu) | 27.6 | 89/118,882/2/0.0007486 | no | 2 | PM3, PP3, BS2 | US | |
| OTOGL | AR | Het | c.2833C>T | p.(Arg945*) | NA | 9/118,122/0/0.00007619 | no | 2 | PVS1, PM3 | P | |
| SYTL4 | XL | Hem | c.164G>T | p.(Ser55Ile) | 26 | 0/121,412/0/0 | RAB8A; RAB27A [32] | no | 2 | PP3, PM2 | US |
| TMED7 | AR | Hom | c.418C>T | p.(Arg140*) | 37 | 0/121,412/0/0 | no | 2 | PP1, PP3, PVS1, PM2 | P | |
| GPR45 | AR | Hom | c.4G>A | p.(Ala2Thr) | 13.47 | 0/121,412/0/0 | no | 1 | PP1, PM2 | US | |
| ID/Gender /Origin | Age at Diagnosis/Age Recent Examination (yrs) | History | Visual Acuity (LogMAR) RE/LE | Refraction | Ophthalmoscopy | Full Field Electroretinogram | Goldmann Perimetry | Optical Coherence Tomography | Fundus Autofluorescence | Other Symptoms |
|---|---|---|---|---|---|---|---|---|---|---|
| A-II:1, ♂ Ghana | 42/58 | Impaired visual acuity and night blindness in 5th decade | CF/CF | RE:S−1.00 C1.25 × 88°/ LE: S−1.25 C1.00 × 89° | Pale optic disks, mild attenuated retinal vessels, extensive atrophy of the RPE in the macula and midperiphery with hyperpigmentations, peripheral RPE spared | NA | Constricted up to 5° | Atrophy of the outer segments | NA | Lens sclerosis, Alpha-thalassemia |
| B-II:1, ♀ The Netherlands | 38/69 | Night blindness in 3rd decade and visual field loss in the 4th decade | 0.15/0.7 | RE: S−1.75 C1.25 × 10°/ LE: S+0.50 C1.25 × 0° | Pale optic disks, severely attenuated retinal vessels, RPE alterations in the posterior pole, midperipheral RPE atrophy and peripheral bonespicule-like pigmentations | Photopic: severely reduced response, Scotopic: no reponse | Constricted up to 10° | Central and peripheral atrophy of the outer segments, intraretinal cysts | Hyper-autofluorescent macula with hypo-autofluorescent perifoveal ring and hypo-autofluoresent, patchy dots | Pseudophakia. RE traumatic corneal perforation and partial aniridia |
| C-II:2, ♀ Pakistan | 20-30/45 | Adult onset nyctalopia and reduced vision, followed by loss of acuity | 0.2/0.3 | Emmetropic | White dots and intra-retinal pigment migration in mid-peripheral retina, especially above and below the optic disc. Bulls eye maculopathy | Rod-cone dysfunction | confrontation approximately 10° | Outer retinal degeneration, relative sparing of fovea | Concentric rings of hypo- and hyper-autofluoresence in the macula, consistent with a bulls eye maculopathy, and areas of hypo-autofluorescence in the mid-periperal retina | - |
| D-II:1, ♀ Muslim Arab | Congenital/37 | Congenital photophobia and reduced visual acuity, congenital nystagmus | 0.1/CF 1m | NA | NA | Photopic: severely reduced responses, Scotopic: slightly reduced responses | NA | NA | NA | - |
| D-II:2, ♀ Muslim Arab | Congenital/25 | Congenital photophobia and reduced visual acuity, congenital nystagmus | CF 2m/CF 2m | NA | NA | Photopic: severely reduced responses, Scotopic: slightly reduced responses | NA | NA | NA | - |
| E-II:3, ♀ Turkey | early infancy/4 | Pendular nystagmus, reduced vision | 1/4 APK/ 1/3 APK | RE: S+4.75 C−2.50 × 2°/LE: S+5.0 C−2.5 × 4° | no visible abnormalities | Photopic: reduced responses, Scotopic: NA | NA | NA | NA | - |
| F-II:2, ♂ Algeria | 2/22 | Night blindness at 2 yrs, at 12 yrs minimal peripheral visual field, moderate photophobia | 1.0/1.0 | RE: S−0.50 C−0.50 × 55°)/LE: C−1.00 × 100°) | Depigmented peripheral retina with bone spicule pigmentary deposits in mid periphery. Macular reflex is normal. Narrowed retinal vessels and pale optic discs LE: interpapillomacular region is depigmented. | NA | Visual field is tubular, with the peripheral isopter V4e shortened at 15-20° around fixative point | CME with foveal thickness of 280 µm RE and 220 µm LE. Outer retinal layers are absent outside the fovea | Typical hypoautofluorescent spots in mid periphery towards the macula. A narrowed ring of hyperautofluorescence around the fovea. LE: hypoautofluorescent spots superonasal of the macula | - |
| G-III:4, ♂ Pakistan | early infancy/15 | Night blindness and visual field loss | 1.0/1.0 | NA | Attenuated retinal vessels, peripheral bone spicule pigmentation | NA | NA | NA | NA | - |
| H-II:1, ♂ The Netherlands | 16/37 | Impaired visual acuity and visual field loss in the 2nd decade | NLP/HM 3m | RE: NA / LE: S−3.00 C1.75 × 177° | RE: phthisis bulbi, LE: Pale optic disk, attenuated retinal vessels, small island with remaining RPE in the macula, extensive atrophy and bone spicule pigmentations in the periphery | Photopic: moderately reduced response, Scotopic: severely reduced responses | LE: Peripheral island inferior | Disorganization and extensive atrophy of the outer retinal layers and thickened RPE in the fovea | NA | RE: Traumatic retinal detachments, aphakia. Proliferative vitreoretinopathy |
| I-IV:2, ♀ Asia | 1st decade/10 | Reduced visual acuity, nystagmus | 0.1/0.1 | NA | Bull’s eye maculopathy | NA | NA | NA | NA | Developmental delay, tonic clonic seizures |
| I-V:1, ♂ Asia | 1st decade/12 | Reduced visual acuity, nystagmus | 0.2/0.3 | NA | Bull’s eye maculopathy | NA | NA | NA | NA | Developmental delay, tonic clonic seizures |
| J-IV:3, ♂ France | 5-20/63 | Night blindness and visual field loss | LP to 1.0/ LP to 1.0 | Variable | Pigment deposits | No or reduced responses | Slightly decreased V4e isopter in periphery to tubular visual field | Moderate decrease of the outer nuclear layer in periphery to complete absence of outer nuclear layer | Typical hypoautofluorescence spots in retinal periphery | - |
| K-IV:1, ♀ The Netherlands | 41/53 | Impaired visual acuity, and night blindness in the 5th decade | 0.7/0.6 | RE: S+2.50 C0.75 × 172° /LE: S+3.00 C1.50 × 165° | Mild pallor of the optic disk, mild attenuated retinal vessels, cystoid maculopathy, thinning of the RPE in the periphery with intraretinal bone spicule pigmentations (LE > RE) | Photopic: severely reduced responses, Scotopic: severely reduced responses | Peripheral sensitivity loss, no absolute defects | CME, peripheral atrophy of the outer segments | Hypoautofluorescent flecks in the fovea, hyperautofluorescent ring along the macula superimposed to the underlying edema, coarse hypoautofluorescent spots along the vascular arcades | Cystoid maculopathy, refractory to systemic treatment with Diamox® and Octreotide® |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Astuti, G.D.N.; Van den Born, L.I.; Khan, M.I.; Hamel, C.P.; Bocquet, B.; Manes, G.; Quinodoz, M.; Ali, M.; Toomes, C.; McKibbin, M.; et al. Identification of Inherited Retinal Disease-Associated Genetic Variants in 11 Candidate Genes. Genes 2018, 9, 21. https://doi.org/10.3390/genes9010021
Astuti GDN, Van den Born LI, Khan MI, Hamel CP, Bocquet B, Manes G, Quinodoz M, Ali M, Toomes C, McKibbin M, et al. Identification of Inherited Retinal Disease-Associated Genetic Variants in 11 Candidate Genes. Genes. 2018; 9(1):21. https://doi.org/10.3390/genes9010021
Chicago/Turabian StyleAstuti, Galuh D. N., L. Ingeborgh Van den Born, M. Imran Khan, Christian P. Hamel, Béatrice Bocquet, Gaël Manes, Mathieu Quinodoz, Manir Ali, Carmel Toomes, Martin McKibbin, and et al. 2018. "Identification of Inherited Retinal Disease-Associated Genetic Variants in 11 Candidate Genes" Genes 9, no. 1: 21. https://doi.org/10.3390/genes9010021
APA StyleAstuti, G. D. N., Van den Born, L. I., Khan, M. I., Hamel, C. P., Bocquet, B., Manes, G., Quinodoz, M., Ali, M., Toomes, C., McKibbin, M., El-Asrag, M. E., Haer-Wigman, L., Inglehearn, C. F., Black, G. C. M., Hoyng, C. B., Cremers, F. P. M., & Roosing, S. (2018). Identification of Inherited Retinal Disease-Associated Genetic Variants in 11 Candidate Genes. Genes, 9(1), 21. https://doi.org/10.3390/genes9010021