Isoform Sequencing and State-of-Art Applications for Unravelling Complexity of Plant Transcriptomes



Abstract
:1. Introduction
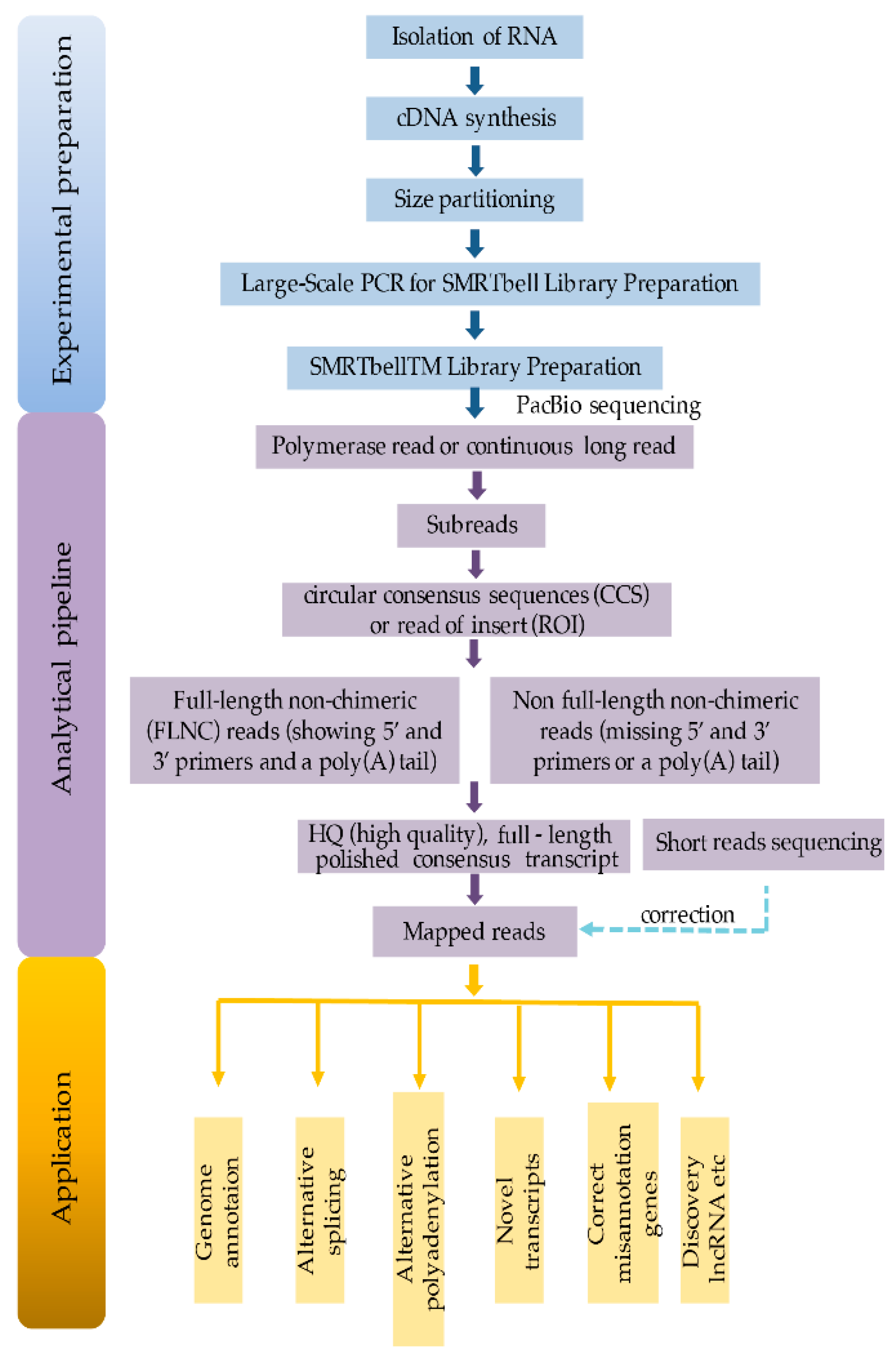
2. Sample Preparation and Library Construction for Isoform Sequencing
2.1. Isolation of Total RNA
2.2. cDNA Synthesis
2.3. Size Partitioning
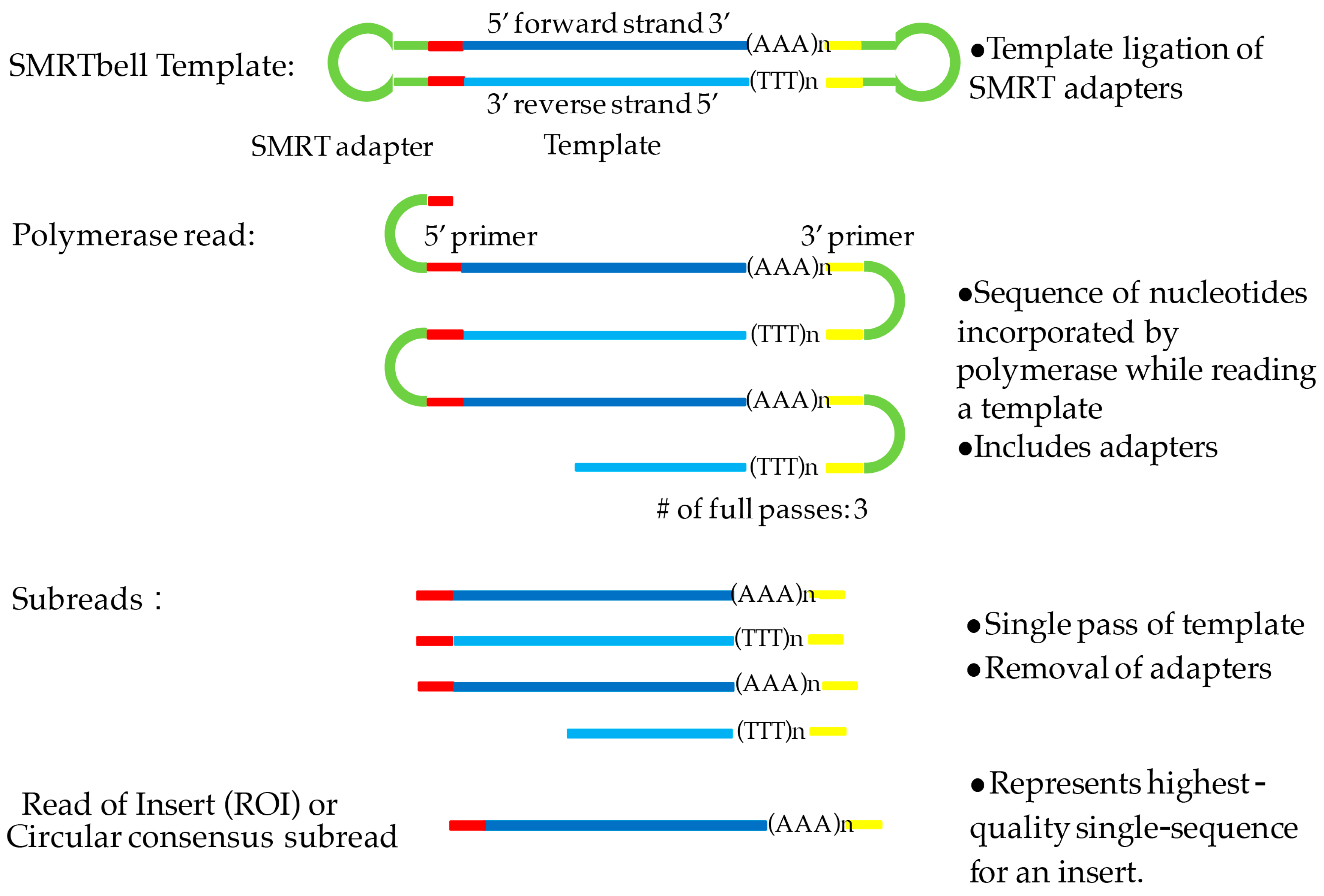
2.4. Library Preparation
3. Bioinformatic Analysis
4. Applications in Plant Transcriptome Research
- (1)
- (2)
- Iso-Seq can generate full-length transcripts, which is fundamental to a newly sequenced genome. It provides golden evidence via alignment against genome to direct delimitate exons, splice sites, and alternative splicing junctions. The continuous sequences guarantee the better accuracy of gene annotations compared to expressed sequence tag (EST), RNA-Seq, and homology inference [30].
- (3)
4.1. Genome Annotation
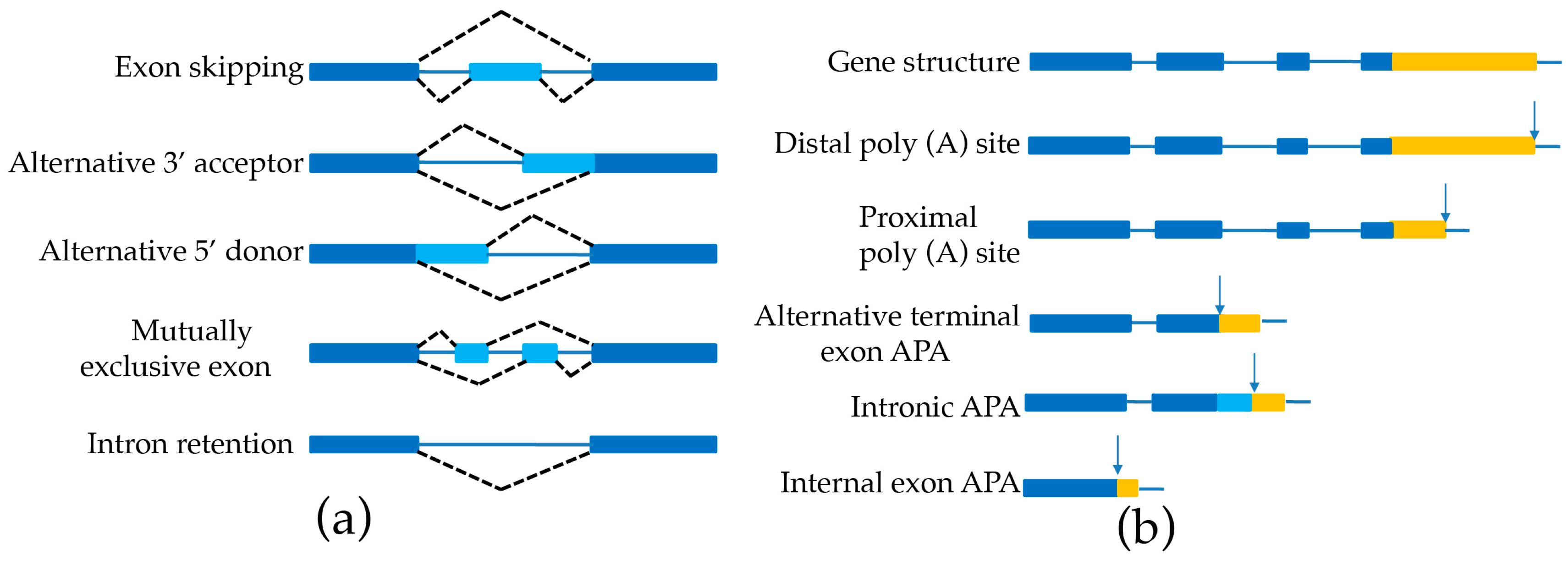
4.2. Alternative Splicing and Alternative Polyadenylation Discovery
4.3. Fusion Genes Determination
4.4. Methylation Detection
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Rhoads, A.; Au, K.F. Pacbio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Travers, K.J.; Chin, C.S.; Rank, D.R.; Eid, J.S.; Turner, S.W. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010, 38, e159. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garay, M.L. Introduction to Isoform Sequencing Using Pacific Biosciences Technology (Iso-Seq); Springer: Dordrecht, The Netherlands, 2015; Volume 9, pp. 141–160. [Google Scholar]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Swarbreck, D.; Wilks, C.; Lamesch, P.; Berardini, T.Z.; Garcia-Hernandez, M.; Foerster, H.; Li, D.; Meyer, T.; Muller, R.; Ploetz, L.; et al. The arabidopsis information resource (tair): Gene structure and function annotation. Nucleic Acids Res. 2008, 36, D1009–D1014. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Zhu, W.; Hamilton, J.; Lin, H.; Campbell, M.; Childs, K.; Thibaud-Nissen, F.; Malek, R.L.; Lee, Y.; Zheng, L.; et al. The TIGR rice genome annotation resource: Improvements and new features. Nucleic Acids Res. 2007, 35, D883–D887. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The b73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- PacBio RS II System. Available online: http://dnatech.genomecenter.ucdavis.edu/pacbio-library-prep-sequencing (accessed on 1 November 2017).
- PacBio Sequel System. Available online: http://www.pacb.com/products-and-services/pacbio-systems/sequel (accessed on 12 July 2017).
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D.; et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012, 30, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, W.; Liu, S.; Schnable, J.C.; Yeh, C.T.; Springer, N.M.; Schnable, P.S.; Barbazuk, W.B. A comprehensive analysis of alternative splicing in paleopolyploid maize. Front. Plant Sci. 2017, 8, 694. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global identification of alternative splicing via comparative analysis of smrt- and illumina-based RNA-SEQ in strawberry. Plant J. Cell Mol. Biol. 2017, 90, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Liu, H.; Zhang, J.; Yang, S.; Kong, G.; Chu, J.S.C.; Chen, N.; Wang, D. Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genom. 2015, 16, 1039. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, H.; Cai, D.; Gao, Y.; Zhang, H.; Wang, Y.; Lin, C.; Ma, L.; Gu, L. Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 2017, 7, 11706. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of salvia miltiorrhizaand tanshinone biosynthesis. Plant J. Cell Mol. Biol. 2015, 82, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L.; et al. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2018, 217, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef] [PubMed]
- Minoche, A.E.; Dohm, J.C.; Schneider, J.; Holtgräwe, D.; Viehöver, P.; Montfort, M.; Sörensen, T.R.; Weisshaar, B.; Himmelbauer, H. Exploiting single-molecule transcript sequencing for eukaryotic gene prediction. Genome Biol. 2015, 16, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, B.; Furtado, A.; Henry, R.J. Long-read sequencing of the coffee bean transcriptome reveals the diversity of full-length transcripts. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- PacBio SMRTbell Library Construction. Available online: http://www.pacb.com/products-and-services/analytical-software/devnet (accessed on 10 May 2017).
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef] [PubMed]
- Au, K.F.; Underwood, J.G.; Lee, L.; Wong, W.H. Improving pacbio long read accuracy by short read alignment. PLoS ONE 2012, 7, e46679. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Rivals, E. Lordec: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackl, T.; Hedrich, R.; Schultz, J.; Förster, F. Proovread: Large-scale high-accuracy pacbio correction through iterative short read consensus. Bioinformatics 2014, 30, 3004–3011. [Google Scholar] [CrossRef] [PubMed]
- Pipelines of TAPIS. Available online: https://bitbucket.org/comp_bio/tapis (accessed on 30 August 2017).
- Pipeline for Iso-Seq. Available online: https://github.com/nextomics/pipeline-for-isoseq (accessed on 24 December 2016).
- Jarvis, D.E.; Ho, Y.S.; Lightfoot, D.J.; Schmockel, S.M.; Li, B.; Borm, T.J.; Ohyanagi, H.; Mineta, K.; Michell, C.T.; Saber, N.; et al. The genome of chenopodium quinoa. Nature 2017, 542, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. Gmap: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-B.; Brendel, V. Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. USA 2006, 103, 7175–7180. [Google Scholar] [CrossRef] [PubMed]
- Foissac, S.; Sammeth, M. Analysis of alternative splicing events in custom gene datasets by astalavista. Methods Mol. Biol. 2015, 1269, 379–392. [Google Scholar] [PubMed]
- Rogers, M.F.; Thomas, J.; Reddy, A.S.; Ben-Hur, A. Splicegrapher: Detecting patterns of alternative splicing from RNA-SEQ data in the context of gene models and EST data. Genome Biol. 2012, 13, R4. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zong, J.; Wei, N.; Cheng, J.; Zhou, X.; Cheng, Y.; Chen, D.; Guo, Q.; Zhang, B.; Feng, Y. Cash: A constructing comprehensive splice site method for detecting alternative splicing events. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Foissac, S.; Sammeth, M. Astalavista: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, W297–W299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-D.; Wang, P.; Bao, Z.; Ma, Q.; Duan, L.-J.; Bao, A.-K.; Zhang, J.-L.; Wang, S.-M. SOS1, HKT1;5, and NHX1 synergistically modulate Na(+) homeostasis in the halophytic grass Puccinellia tenuiflora. Front. Plant Sci. 2017, 8, 576. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.N. Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol. 2007, 58, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Li, Q.Q. Alternative polyadenylation and gene expression regulation in plants. Wiley Interdiscip. Rev. RNA 2010, 2, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, M.; Downie, B.; Liang, C.; Ji, G.; Li, Q.Q.; Hunt, A.G. Genome-wide landscape of polyadenylation in arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. USA 2011, 108, 12533–12538. [Google Scholar] [CrossRef] [PubMed]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Venu, R.C.; Nobuta, K.; Wu, X.; Notibala, V.; Demirci, C.; Meyers, B.C.; Wang, G.L.; Ji, G.; Li, Q.Q. Transcriptome dynamics through alternative polyadenylation in developmental and environmental responses in plants revealed by deep sequencing. Genome Res. 2011, 21, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Ji, G.; Haas, B.J.; Wu, X.; Zheng, J.; Reese, G.J.; Li, Q.Q. Genome level analysis of rice mRNA 3′-end processing signals and alternative polyadenylation. Nucleic Acids Res. 2008, 36, 3150–3161. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Marquardt, S.; Lister, C.; Swiezewski, S.; Dean, C. Targeted 3’ processing of antisense transcripts triggers arabidopsis FLC chromatin silencing. Science 2010, 327, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Addepalli, B.; Yun, K.-Y.; Hunt, A.G.; Xu, R.; Rao, S.; Li, Q.Q.; Falcone, D.L. A polyadenylation factor subunit implicated in regulating oxidative signaling in arabidopsis thaliana. PLoS ONE 2008, 3, e2410. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, K.; Cross, J.M. Genetic approaches to study plant responses to environmental stresses: An overview. Biology 2016, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Meyer, C.A.; Wang, Q.; Liu, J.S.; Shirley Liu, X.; Zhang, Y. GFOLD: A generalized fold change for ranking differentially expressed genes from rna-seq data. Bioinformatics 2012, 28, 2782–2788. [Google Scholar] [CrossRef] [PubMed]
- Tardaguila, M.; de la Fuente, L.; Marti, C.; Pereira, C.; del Risco, H.; Ferrell, M.; Mellado, M.; Macchietto, M.; Verheggen, K.; Edelmann, M.; et al. SQANTI: Extensive characterization of long read transcript sequences for quality control in full-length transcriptome identification and quantification. BioArxiv 2017. [Google Scholar] [CrossRef]
- Weirather, J.L.; Afshar, P.T.; Clark, T.A.; Tseng, E.; Powers, L.S.; Underwood, J.G.; Zabner, J.; Korlach, J.; Wong, W.H.; Au, K.F. Characterization of fusion genes and the significantly expressed fusion isoforms in breast cancer by hybrid sequencing. Nucleic Acids Res. 2015, 43, e116. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Tang, Z.; Wang, M.; Gao, W.; Tu, L.; Jin, X.; Chen, L.; He, Y.; Zhang, L.; Zhu, L.; et al. The genome sequence of sea-island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 2015, 5, 17662. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, J.A.; Govindarajulu, R.; Ashman, T.L.; Liston, A. Evolutionary origins and dynamics of octoploid strawberry subgenomes revealed by dense targeted capture linkage maps. Genome Biol. Evol. 2014, 6, 3295–3313. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xiao, G.; Zhu, Y.X. Single-nucleotide resolution mapping of the Gossypium raimondii transcriptome reveals a new mechanism for alternative splicing of introns. Mol. Plant 2014, 7, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Amborella Genome Database. Available online: http://amborella.huck.psu.edu/ (accessed on 11 March 2017).
- Seymour, D.K.; Becker, C. The causes and consequences of DNA methylome variation in plants. Curr. Opin. Plant Biol. 2017, 36, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, N.D.; Karri, S.; Fang, G.; Bashir, A. Detecting epigenetic motifs in low coverage and metagenomics settings. BMC Bioinform. 2014, 15, S16. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Van Bel, M.; Woloszynska, M.; Slabbinck, B.; Martens, C.; De Block, M.; Coppens, F.; Van Lijsebettens, M. Plant-RRBS, a bisulfite and next-generation sequencing-based methylome profiling method enriching for coverage of cytosine positions. BMC Plant Biol. 2017, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Species | Sample Collection | RNA Extraction | Size-Fractionated Libraries | Platform and Throughput | Ref. |
|---|---|---|---|---|---|
| sorghum (BTx623) | Control and drought treatment of 7-day-old seedlings for 6 h | TRIzol reagent (Invitrogen, Carlsbad, CA, USA) with DNaseI (Fermentas, Waltham, MA, USA) | 1–2 kb and 2–6 kb | PacBio RS II with 28 SMRT cells | [16] |
| maize B73 | Root, pollen, embryo, endosperm, immature ear and immature tassel | TRIzol reagent (Invitrogen, Carlsbad, CA, USA) with RQ1 DNase (Promega, Madison, WI, USA) | <1 kb, 1–2 kb, 2–3 kb, 3–5 kb, 4–6 kb and >5 kb | PacBio RS II with 47 SMRT cells | [13] |
| wheat Xiaoyan | Unfertilized caryopses and developing grains | RNA extraction kit (Takara Biotechnology, Dalian, Liaoning, China) with TURBO DNaseI (Promega, Madison, WI, USA) | <2 kb, ≥2 kb | PacBio RS II with 8 SMRT cells | [14] |
| Amborella trichopoda | Young leaves and female flowers | CTAB method and RNeasy Mini extraction kit (Qiagen, Hilden, Germany) with TURBO DNA-free Kit | 1–2 kb, 2–3 kb and >3 kb | PacBio RS II with 19 SMRT cells | [17] |
| wild strawberry | Receptacle of five different stages | Plant Total RNA Isolation Kit (Sangon Biotech, Shanghai, China) | 1–2 kb, 2–3 kb and >3 kb | PacBio RS with 13 SMRT cells | [12] |
| moso bamboo | Underground rhizome, lateral rhizome, shoot, root, and leaf | RNAprep Pure Plant Kit (Tiangen, Beijing, China) with DNase I | 1–2 kb, 2–3 kb and >3 kb | PacBio RS II with 7 SMRT cells | [15] |
| Salvia miltiorrhiza | Periderm, phloem, and xylem from roots | RNeasy Plus Mini Kit (#74134, Qiagen, Hilden, Germany) | <1 kb, 1–2 kb, 2–3 kb and >3 kb | PacBio RS with 8 SMRT cells | [18] |
| cotton | Root, hypocotyl, leaf, petal, anther, stigma; fibre samples | Spectrum Plant Total RNA kit (Sigma-Aldrich, St. Louis, MI, USA) | 1–2 kb, 2–3 kb and 3–6 kb | PacBio RS II with 30 SMRT cells | [19] |
| sugarcane | Leaf, internode, and root tissues of different stages | TRIzol (Invitrogen) and Qiagen RNeasy Plant minikit (#74134, Qiagen, Hilden, Germany) | 0.5–2.5 kb, 2–3.5 kb, 3–6 kb and 5–10 kb | PacBio RS II with 6 SMRT cells | [20] |
| sugar beet | Seedlings | Nucleospin Plant RNA kit (Macherey-Nagel, Duren, Germany) | 1–2 kb, 2–3 kb and >3 kb | PacBio RS with 6 SMRT cells | [21] |
| coffee bean | Immature, intermediated, and mature fruits | TRIzol plus RNA purification kit (Invitrogen, Carlsbad, CA, USA), the RNeasy Plant Mini Kit (#74903, Qiagen, Hilden, Germany) | 0.5–2.5 kb, 2–3.5 kb, 3–6 kb and 5–10 kb | PacBio RS II with 2 SMRT cells | [22] |
| Species | ROI | Full-Length ROI | Error Correction FLNC Reads | Mapped Reads |
|---|---|---|---|---|
| sorghum (BTx623) | 1,838,330 | 884,638 | NA | 867,089 |
| maize B73 | 3,716,604 | 1,553,692 | 643,330 | 606,145 |
| wheat Xiaoyan | 240,312 | NA | 197,709 | 91,881 |
| Amborella trichopoda | 660,458 | 217,954 | 146,686 1 | 124,509 2 |
| wild strawberry | 442,601 | 354,393 | 85,416 | 82,360 |
| moso bamboo | 288,312 | 147,362 | 146,225 | 145,522 |
| Salvia miltiorrhiza | 796,011 | 223,368 | NS | NA |
| cotton | 2,542,318 | 1,096,932 | NA | 339,230 |
| sugar cane | 290,393 | 186,999 | 107,604 | 74,716 |
| sugar beet | 395,038 | 109,920 | NA | 107,721 |
| coffee bean | 433,877 | 233,464 | NA | NA |
| Species | Isoform | Novel Transcripts | AS | APA | Novel Genes | lncRNA | Mis-Annotated Genes |
|---|---|---|---|---|---|---|---|
| sorghum (BTx623) | 27,860 | 11,342 | 10,053 | 11,013 | 2171 | 540 | 941 |
| maize B73 | 111,151 | 65,350 | NS | NA | 2253 | 867 | 2199 * |
| wheat Xiaoyan | 22,768 | 9591 | NS | NA | 3026 | NA | 180 |
| Amborella trichopoda | 10,617 | 3680 | 4879 | NA | 510 | NA | 3255 |
| wild strawberry | 33,236 | 5501 | 17,260 | NA | 3649 | NA | NA |
| moso bamboo | 42,280 | 35,447 | 21,154 | 6311 | 8091 | 3096 | 2241 |
| Salvia miltiorrhiza | 160,468 | NA | 4165 | NA | NA | 11,046 | NA |
| cotton | 176,849 | 13,551 | 133,329 | 43,784 | NA | 2447 | NA |
| sugar cane | 107,598 | 2450 | 4870 | NA | NA | 2426 | NA |
| sugar beet | NA | NA | NA | NA | NA | NA | 4000 |
| coffee bean | 95,995 | NA | NS | NS | 1213 | NA | NA |
| Species | Read Processing | Correction | Mapping | AS | Novel Gene | APA |
|---|---|---|---|---|---|---|
| sorghum (BTx623) | TAPIS | LoRDEC, proovread and TAPIS | GMAP | SpliceGrapher | TAPIS | TAPIS |
| maize B73 | ToFU | ICE-Quiver | GMAP | AStalavista | BLASTN | NA |
| wheat Xiaoyan | SMRT analysis | SMRT analysis, proovread | GMAP | In-house perl script | GMAP | NA |
| Amborella trichopoda | SMRT analysis_v2.2.0 | minFullPasses, LSC-corrected and ICE-Quiver | GMAP, BLAT | PASA, de novo AS detection | NA | NA |
| wild strawberry | RS_IsoSeq_v2.3 | ICE-Quiver, LoRDEC | GMAP | AStalavista | NA | NA |
| moso bamboo | SMRT analysis_2.3.0 | LSC | GMAP | AStalavista | TAPIS | TAPIS |
| Salvia miltiorrhiza | SMRT analysis_2.2.0 | LSC | GMAP | SPLICEMAP | SPLICEMAP | NA |
| cotton | SMRT analysis | pipeline-for-Iso-Seq | GMAP | alternative_splice.py | BLAST | SMRT analysis |
| sugar cane | SMRT analysis_2.3.0 | ICE-Quiver, proovread, and LoRDEC | GMAP | TAPIS | BLAST | NA |
| sugar beet | SMRT analysis_v2.0 | Proovread, normalize-by-median.py | GMAP, AUGUSTUS | NA | NA | NA |
| coffee bean | RS_IsoSeq_v2.3 | ICE-Quiver | BLAST | BLAST | BLAST | BLAST |
| Species | Iso-Seq | SGS/Sanger | Reference | |
|---|---|---|---|---|
| Isoform number per gene | cotton | 3.93 | 1.35 | [19,51] |
| maize B73 | 6.56 * | 2.84 * | [13] | |
| Total isoform number | wild strawberry | 26,676 | 20,705 | [12] |
| moso bamboo | 42,280 | 10,471 | [15] | |
| Average gene length (bp) | Amborella trichopoda | 2044 | 950 1 | [17] |
| maize B73 | 2632 | 1684 | [7,13] | |
| wild strawberry | 2466 | 1187 | [12,52] | |
| cotton | 2175 | 1462 | [19,51] | |
| AS events | wild strawberry | 17,260 | 12,080 | [12] |
| cotton | 133,229 | 16,437 | [19,53] | |
| Number of fusion genes | maize B73 | 1430 | 134 | [13] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, D.; Cao, H.X.; Li, C.; Humbeck, K.; Wang, W. Isoform Sequencing and State-of-Art Applications for Unravelling Complexity of Plant Transcriptomes. Genes 2018, 9, 43. https://doi.org/10.3390/genes9010043
An D, Cao HX, Li C, Humbeck K, Wang W. Isoform Sequencing and State-of-Art Applications for Unravelling Complexity of Plant Transcriptomes. Genes. 2018; 9(1):43. https://doi.org/10.3390/genes9010043
Chicago/Turabian StyleAn, Dong, Hieu X. Cao, Changsheng Li, Klaus Humbeck, and Wenqin Wang. 2018. "Isoform Sequencing and State-of-Art Applications for Unravelling Complexity of Plant Transcriptomes" Genes 9, no. 1: 43. https://doi.org/10.3390/genes9010043
APA StyleAn, D., Cao, H. X., Li, C., Humbeck, K., & Wang, W. (2018). Isoform Sequencing and State-of-Art Applications for Unravelling Complexity of Plant Transcriptomes. Genes, 9(1), 43. https://doi.org/10.3390/genes9010043