Clinical Implications of Hepatitis B Virus RNA and Covalently Closed Circular DNA in Monitoring Patients with Chronic Hepatitis B Today with a Gaze into the Future: The Field Is Unprepared for a Sterilizing Cure



Abstract
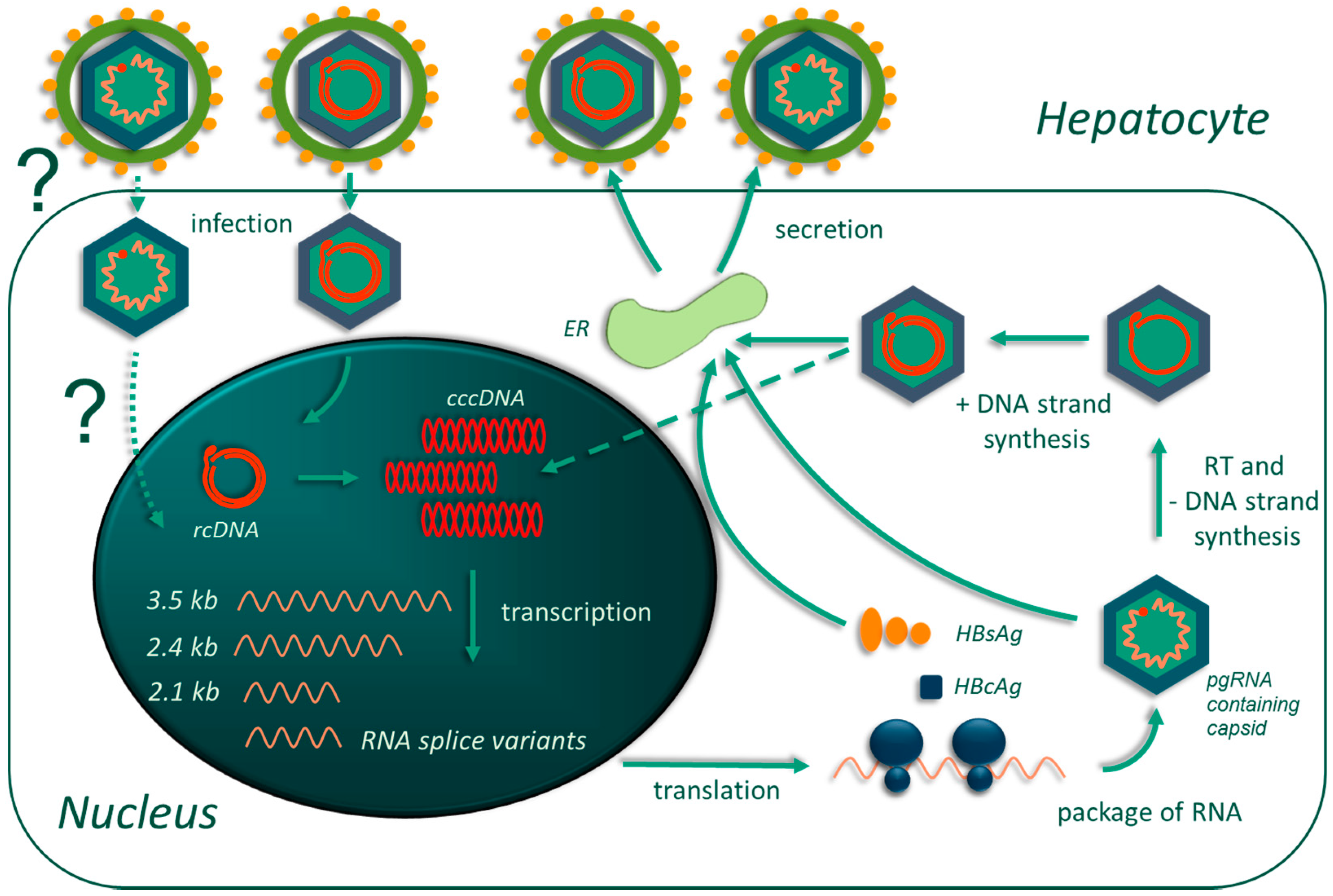
:1. Introduction
2. Fundamental Pathobiology of Hepatitis B Virus
3. Chronic Hepatitis B: Chronicity, Clinical Course, and Clinical Markers
4. Therapeutic Options for Chronic Hepatitis B: Raiders of the Deceptive Cure
5. Covalently Closed Circular DNA Levels: A Marker for Predicting Antiviral Response
6. Covalently Closed Circular DNA and Adverse Chronic Hepatitis B Outcomes
7. Hepatitis B Virus RNA in Secreted Virions
8. Hepatitis B Virus RNA Splice Variants in Disease Progression and Outcomes
9. Methods and Challenges of Detecting cccDNA and pgRNA
10. Conclusions
Funding
Conflicts of Interest
References
- Dandri, M.; Locarnini, S. New insight in the pathobiology of hepatitis B virus infection. Gut 2012, 61, i6–i17. [Google Scholar] [CrossRef] [PubMed]
- Glebe, D.; Bremer, C.M. The molecular virology of hepatitis B virus. Semin. Liver Dis. 2013, 33, 103–112. [Google Scholar] [PubMed]
- Xie, Y. Hepatitis B virus-associated hepatocellular carcinoma. Adv. Exp. Med. Biol. 2017, 1018, 11–21. [Google Scholar] [PubMed]
- Papatheodoridis, G.V.; Chan, H.L.-Y.; Hansen, B.E.; Janssen, H.L.A.; Lampertico, P. Risk of hepatocellular carcinoma in chronic hepatitis B: Assessment and modification with current antiviral therapy. J. Hepatol. 2015, 62, 956–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.-C.; Chen, C.-H.; Hu, T.-H.; Lu, S.-N.; Lee, C.-M.; Wang, J.-H.; Hung, C.-H. Long-term outcomes of hepatitis B virus-related cirrhosis treated with nucleos(t)ide analogs. J. Formos. Med. Assoc. 2017, 116, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Razavi-Shearer, D.; Gamkrelidze, I.; Nguyen, M.H.; Chen, D.S.; van Damme, P.; Abbas, Z.; Abdulla, M.; Abou Rached, A.; Adda, D.; Aho, I.; et al. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef]
- GBO 2016 Causes of Death Collaborators. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Venook, A.P.; Papandreou, C.; Furuse, J.; de Guevara, L.L. The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. Oncologist 2010, 15, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Zhang, X.-Y. Innate immune recognition of hepatitis B virus. World J. Hepatol. 2015, 7, 2319–2322. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.-C.; Huang, L.R. Immunopathogenesis of hepatitis B virus. J. Infect. Dis. 2017, 216, S765–S770. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Xia, Y.; Serti, E.; Block, P.D.; Chung, M.; Chayama, K.; Rehermann, B.; Liang, T.J. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 2017, 66, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Suslov, A.; Boldanova, T.; Wang, X.; Wieland, S.; Heim, M.H. Hepatitis B virus does not interfere with innate immune responses in the human liver. Gastroenterology 2018, 154, 1778–1790. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Cjakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus (HBV) genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, Y.; Li, G.; Shen, C.; Li, J.; Chen, S.; Zhang, X.; Zhu, M.; Zheng, J.; Song, Z.; et al. Natural history of serum HBV-RNA in chronic HBV infection. J. Viral Hepat. 2018, 25, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Allweiss, L.; Volz, T.; Dandri, M.; Lutgehetmann, M. Serum HBV pgRNA as a clinical marker for cccDNA activity. J. Hepatol. 2017, 66, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA integration: Molecular mechanisms and clinical implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B virus DNA integration occurs early in the viral life cycle in an in vitro infection model via NTCP-dependent uptake of enveloped virus particles. J. Virol. 2018, JVI-02007. [Google Scholar] [CrossRef]
- Saitta, C.; Tripodi, G.; Barbera, A.; Bertuccio, A.; Smedile, A.; Ciancio, A.; Raffa, G.; Sangiovanni, A.; Navarra, G.; Raimondo, G.; et al. Hepatitis B virus (HBV) DNA integration in patients with occult HBV infection and hepatocellular carcinoma. Liver Int. 2015, 35, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Busca, A.; Kumar, A. Innate immune responses in hepatitis B virus (HBV) infection. Virol. J. 2014, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faure-Dupuy, S.; Lucifora, J.; Durantel, D. Interplay between the hepatitis B virus and innate immunity: From an understanding to the development of therapeutic concepts. Viruses 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Fung, J.; Lai, C.-L.; Seto, W.-K.; Yuen, M.-F. Nucleoside/nucleotide analogues in the treatment of chronic hepatitis B. J. Antimicrob. Chemother. 2011, 66, 2715–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, K.; Brunetto, M.; Seto, W.K.; Lim, Y.S.; Fung, S.; Marcellin, P.; Ahn, S.H.; Izumi, N.; Chuang, W.L.; Bae, H.; et al. 96 weeks treatment of tenofovir alafenamide vs. tenofovir disoproxil fumarate for hepatitis B virus infection. J. Hepatol. 2018, 68, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-W.; Chayama, K.; Tsuge, M.; Takahashi, S.; Hatakeyama, T.; Abe, H.; Hu, J.T.; Liu, C.J.; Lai, M.Y.; Chen, D.S.; et al. Differential effects of interferon and lamivudine on serum HBV RNA inhibition in patients with chronic hepatitis B. Antivir. Ther. 2010, 15, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Rijckborst, V.; Janssen, H.L.A. The role of interferon in hepatitis B therapy. Curr. Hepat. Rep. 2010, 9, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, A.S.J.; Kwok, R.; Ahmed, T. Alpha-interferon treatment in hepatitis B. Ann. Transl. Med. 2017, 5, 159. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, F.; Arase, Y.; Suzuki, Y.; Akuta, N.; Sezaki, H.; Seko, Y.; Kawamura, Y.; Hosaka, T.; Kobayashi, M.; Saito, S.; et al. Long-term efficacy of interferon therapy in patients with chronic hepatitis B virus unfection in Japan. J. Gastroenterol. 2012, 7, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Rodríguez, C.M.; Gutiérrez-García, M.L. Prevention of hepatocellular carcinoma in patients with chronic hepatitis B. World J. Gastrointest. Pharmacol. Ther. 2014, 5, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Van Bömmel, F.; van Bömmel, A.; Krauel, A.; Wat, C.; Pavlovic, V.; Yang, L.; Deichsel, D.; Berg, T.; Böhm, S. Serum HBV RNA as a predictor of peginterferon alfa-2a (40KD) response in patients with HBeAg-positive chronic hepatitis B. J. Infect. Dis. 2018, 218, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Yang, F.-C.; Chen, Y.; Jiang, X.-Y.; Yan, L.; Jiang, L.-Y.; Gong, J.-P.; Zhang, D.-Z.; Ren, H.; Liao, Y. Baseline value of intrahepatic HBV DNA over cccDNA predicts patient’s response to interferon therapy. Sci. Rep. 2017, 7, 5937. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, M.; Wang, F.; Zhang, W.; Wang, W.; Zhang, X.; Zhang, J.; Liu, Y.; Liu, Y.; Feng, Y.; et al. Hepatitis B virus spliced variants are associated with an impaired response to interferon therapy. Sci. Rep. 2015, 5, 16459. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, F.; Miyakoshi, H.; Kobayashi, M.; Kumada, H. Correlation between serum hepatitis B virus core-related antigen and intrahepatic covalently closed circular DNA in chronic hepatitis B patients. J. Med. Virol. 2009, 81, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.-B.; Zhu, X.; Yan, L.B.; Du, L.Y.; Liu, C.; Liao, J.; Tang, H. Quantitative intrahepatic HBV cccDNA correlates with histological liver inflammation in chronic hepatitis B virus infection. Int. J. Infect. Dis. 2016, 52, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, T.; Noguchi, C.; Hiraga, N.; Mori, N.; Tsuge, M.; Imamura, M.; Takahashi, S.; Kawakami, Y.; Fijumoto, Y.; Ochi, H.; et al. Serum HBV RNA is a predictor of early emergence of the YMDD mutant in patients treated with lamivudine. Hepatology 2007, 45, 1179–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bömmel, F.; Berg, T. Stopping long-term treatment with nucleos(t)ide analogues is a favourable option for selected patients with HBeAg-negative chronic hepatitis B. Liver Int. 2018, 38, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cubero, E.; del Arco, R.T.S.; Peña-Asensio, J.; de Villalobos, E.S.; Míquel, J.; Larrubia, J.R. Is it possible to stop nucleos(t)ide analogue treatment in chronic hepatitis B patients? World J. Gastroenterol. 2018, 24, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, Q.; Zhu, X.; Lin, C.; Chen, Y.; Deng, H.; Mei, Y.; Zhao, Z.; Xie, D.; Gao, Z.; et al. 48-week outcome after cessation of nucleos(t)ide analogue treatment in chronic hepatitis B patient and the associated factors with relapse. Can. J. Gastroenterol. Hepatol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, T.; Huang, X.; Kumar, G.R.; Chen, X.; Zeng, Z.; Zhang, R.; Chen, R.; Li, T.; Zhang, T.; et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J. Hepatol. 2016, 65, 700–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-H.; Hsu, Y.C.; Lu, S.N.; Hung, C.H.; Wang, J.H.; Lee, C.M.; Hu, T.H. The incidence and predictors of HBV relapse after cessation of tenofovir therapy in chronic hepatitis B patients. J. Viral Hepat. 2018, 25, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Tanaka, Y.; Orito, E.; Hirashima, N.; Ide, T.; Hino, T.; Kumashiro, R.; Kato, A.; Nukaya, H.; Sakakibara, K.; et al. Predicting relapse after cessation of lamivudine monotherapy for chronic hepatitis B virus infection. Clin. Infect. Dis. 2004, 38, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.G.; Wai, C.T.; Rajnakova, A.; Kajiji, T.; Guan, R. Fatal hepatitis B reactivation following discontinuation of nucleoside analogues for chronic hepatitis B. Gut 2002, 51, 597–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-C.; Chen, P.-J. The potential and challenges of CRISPR-Cas in eradication of hepatitis B virus covalently closed circular DNA. Virus Res. 2018, 244, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.; Shlomai, A.; Cox, D.B.; Schwartz, R.E.; Michailidis, E.; Bhatta, A.; Scott, D.A.; Zhang, F.; Rice, C.M.; Bhatia, S.N. CRISPR/Cas9 cleavage of viral dna efficiently suppresses hepatitis B virus. Sci. Rep. 2015, 5, 10833. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, R.; Zhang, R.; Ding, S.; Zhang, T.; Yuan, Q.; Guan, G.; Chen, X.; Zhang, T.; Zhuang, H.; et al. The gRNA-miRNA-gRNA ternary cassette combining CRISPR/Cas9 with RNAi approach strongly inhibits hepatitis B virus replication. Theranostics 2017, 7, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Masaki, K.; Abe-Chayama, H.; Mochida, K.; Yamamoto, T.; Chayama, K. Highly multiplexed CRISPR-Cas9-nuclease and Cas9-nickase vectors for inactivation of hepatitis B virus. Genes Cells 2016, 21, 1253–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, C.; Sohn, J.A. Complete spectrum of CRISPR/Cas9-induced mutations on HBV cccDNA. Mol. Ther. 2016, 24, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Xie, K.; Xu, Y.; Wang, L.; Chen, K.; Zhang, L.; Fang, J. CRISPR/Cas9 produces anti-hepatitis B virus effect in hepatoma cells and transgenic mouse. Virus Res. 2016, 217, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Kornepati, A.V.R.; Cullen, B.R. Targeting hepatitis B virus cccDNA using CRISPR/Cas9. Antiviral Res. 2015, 123, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, M.; Gong, M.; Xu, Y.; Xie, C.; Deng, H.; Li, X.; Wu, H.; Wang, Z. Inhibition of hepatitis B virus replication via HBV DNA cleavage by Cas9 from Staphylococcus aureus. Antiviral Res. 2018, 152, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.; Moyo, B.; Nicholson, S.; Maepa, M.B.; Watashi, K.; Ely, A.; Weinberg, M.S.; Arbuthnot, P. ssAAVs containing cassettes encoding SaCas9 and guides targeting hepatitis B virus inactivate replication of the virus in cultured cells. Sci. Rep. 2017, 7, 7401. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzi, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 6176. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hösel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-γ and tumor necrosis factor-α produced by T cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Kostiushev, D.; Brezgin, S.; Kostyusheva, A.; Zarifyan, D.; Chulanov, V. A novel CRISPR/Cas9-based approach to transient activation of intracellular host restriction factors results in strong suppression of hepatitis B virus and degradation of cccDNA. J. Viral Hepatitis 2018, 25, 16–17. [Google Scholar]
- Krebs, K.; Böttingeer, N.; Huang, L.R.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Kruse, R.L.; Shum, T.; Tashiro, H.; Barzi, M.; Yi, Z.; Whitten-Bauer, C.; Legras, X.; Bissig-Choisat, B.; Garaigorta, U.; Gottschalk, S.; et al. HBsAg-redirected T cells exhibit antiviral activity in HBV-infected human liver chimeric mice. Cytotherapy 2018, 20, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, J.; Yuan, Q.; Xia, N. Detection of HBV covalently closed circular DNA. Viruses 2017, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, Y.; Meng, Q.; Zhang, Z.; Zhao, P.; Shang, Q.; Li, Y.; Su, M.; Li, T.; Su, M.; et al. Serum HBV DNA, RNA and HBsAg: Which correlated better to intrahepatic covalently closed circular DNA before and after nucleos(t)ide analogue treatment? J. Clin. Microbiol. 2017, 55, 2972–2982. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.S.; Marion, P.L.; Miller, R.H. The hepadna viruses of animals. Semin. Liver Dis. 1984, 4, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Hu, J. Formation of hepatitis B virus covalently closed circular DNA: Removal of genome-linked protein. J. Virol. 2007, 81, 6164–6174. [Google Scholar] [CrossRef] [PubMed]
- Takkenberg, R.B.; Menting, A.; Beld, M.G.H.M. Validation of a sensitive and specific real-time PCR for detection and quantitation of hepatitis B virus covalently closed circular DNA in plasma of chronic hepatitis B patients. Methods Mol. Biol. 2012, 903, 113–128. [Google Scholar] [PubMed]
- Zhang, X.; Lu, W.; Zheng, Y.; Wang, W.; Bai, L.; Chen, L.; Feng, Y.; Zhang, Z.; Yuan, Z. In situ analysis of intrahepatic virological events in chronic hepatitis B virus infection. J. Clin. Investig. 2016, 126, 1079–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yue, L.; Zhang, Z.; Yuan, Z. Establishment of a fluorescent in situ hybridization assay for imaging hepatitis B virus nucleic acids in cell culture models. Emerg. Microbes Infect. 2017, 6, e98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revill, P.A.; Locarnini, S.A. New perspectives on the hepatitis B virus life cycle in the human liver. J. Clin. Investig. 2016, 126, 833–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W. The hepatitis B virus receptor. Annu. Rev. Cell Dev. Biol. 2015, 31, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Urban, S.; Li, W.; Wakita, T. NTCP and beyond: Opening the door to unveil hepatitis B virus entry. Int. J. Mol. Sci. 2014, 15, 2892–2905. [Google Scholar] [CrossRef] [PubMed]
- König, A.; Döring, B.; Mohr, C.; Geipel, A.; Geyer, J.; Glebe, D. Kinetics of the bile acid transporter and hepatitis B virus receptor Na+/taurocholate cotransporting polypeptide (NTCP) in hepatocytes. J. Hepatol. 2014, 61, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Kullak-Ublick, G.A.; Stieger, B.; Meier, P.J. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology 2004, 126, 322–342. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köck, J.; Rösler, C.; Zhang, J.-J.; Blum, H.E.; Nassal, M.; Thoma, C. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 2010, 6, e1001082. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap endonuclease 1 is involved in cccDNA formation in the hepatitis B virus. PLoS Pathog. 2018, 14, e1007124. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA polymerase κ is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef] [PubMed]
- Königer, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.; Nassal, M. A role for the host DNA damage response in hepatitis B virus cccDNA formation—and beyond? Viruses 2017, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Moreno, A.; Garaigorta, U. Hepatitis B virus and DNA damage response: Interactions and consequences for the infection. Viruses 2017, 9, 304. [Google Scholar] [CrossRef] [PubMed]
- Sommer, G.; van Bömmel, F.; Will, H. Genotype-specific synthesis and secretion of spliced hepatitis B virus genomes in hepatoma cells. Virology 2000, 271, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Seifer, M.; Standring, D.N. A protease-sensitive hinge linking the two domains of the hepatitis B virus core protein is exposed on the viral capsid surface. J. Virol. 1994, 68, 5548–5555. [Google Scholar] [PubMed]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fufe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Ferrari, C.; Pasquinelli, C.; Chisari, F.V. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T–lymphocyte response. Nat. Med. 1996, 2, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Moraleda, G.; Saputelli, J.; Aldrich, C.E.; Averett, D.; Condreay, L.; Mason, W.S. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J. Virol. 1997, 71, 9392–9399. [Google Scholar] [PubMed]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schöneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV bypasses the innate immune response and does not protect HCV from antiviral activity of interferon. Gastroenterology 2018, 154, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.F.; Chisari, F.V. Stealth and cunning: Hepatitis B and hepatitis C viruses. J. Virol. 2005, 79, 9369–9380. [Google Scholar] [CrossRef] [PubMed]
- Pagliaccetti, N.E.; Robek, M.D. Interferon-λ in the immune response to hepatitis B virus and hepatitis C virus. J. Interf. Cytokine Res. 2010, 30, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Robek, M.D.; Boyd, B.S.; Wieland, S.F.; Chisari, F.V. Signal transduction pathways that inhibit hepatitis B virus replication. Proc. Natl. Acad. Sci. USA 2004, 101, 1743–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.-H.; Park, E.S.; Kim, D.H.; Cho, K.C.; Kim, K.P.; Park, Y.K.; Ahn, S.H.; Park, S.H.; Kim, K.H.; Kim, C.W.; et al. Suppression of interferon-mediated anti-HBV response by single CpG methylation in the 5′-UTR of TRIM22. Gut 2018, 67, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, C.; Wang, F.; Zhu, T.; Li, S.; Liu, S.; Xiao, F. Expression of interferon effector gene SART1 correlates with interferon treatment response against hepatitis B infection. Mediators Inflamm. 2016, 2016, 3894816. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Yim, S.A.; Heydmann, S.; El Saghire, H.; Bach, C.; Turon-Lagot, V.; Mailly, L.; Durand, S.C.; Lucifora, J.; Durante, D.; et al. Hepatitis B virus evasion from cyclic guanosine monophosphate-adenosine monophosphate synthase sensing in human hepatocytes. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Peppa, D. NK cells: A double-edged sword in chronic hepatitis B virus infection. Front. Immunol. 2013, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Bi, L.; Zhou, J.; Zhou, D.; Liu, Y.; Jin, G.; Yan, W. Modulation of the function of dendritic cells in adolescents with chronic HBV infection by IFN-λ1. Int. J. Clin. Exp. Pathol. 2015, 8, 1743–1751. [Google Scholar] [PubMed]
- Chisari, F.V.; Isogawa, M.; Wieland, S.F. Pathogenesis of hepatitis B virus infection. Pathol. Biol. 2010, 58, 258–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, A.S.; Zoulim, F.; Dusheiko, G.; Ghany, M.G. Hepatitis B cure: From discovery to regulatory approval. Hepatology 2017, 66, 1296–1313. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, M.; Böhm, S.; Schott, T.; Deichsel, D.; Bremer, C.M.; Schröder, K.; Gerlich, W.H.; Glebe, D.; Berg, T.; van Bömmel, F. Quantification of large and middle proteins of hepatitis B virus surface antigen (HBsAg) as a novel tool for the identification of inactive HBV carriers. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Dandri, M. The role of cccDNA in HBV maintenance. Viruses 2017, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-Y.; Wen, Y.-M. A sandwich strategy for functional cure of chronic hepatitis B. Emerg. Microbes Infect. 2018, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Guo, F.; Zhao, X.; Guo, J.-T. Therapeutic strategies for a functional cure of chronic hepatitis B virus infection. Acta Pharm. Sin. B 2014, 4, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.R. Emerging therapies toward a functional cure for hepatitis B virus infection. Gastroenterol. Hepatol. 2018, 14, 439–442. [Google Scholar]
- Hoofnagle, J.H. Reactivation of hepatitis B. Hepatology 2009, 49, S156–S165. [Google Scholar] [CrossRef] [PubMed]
- Ridruejo, E.; Marciano, S.; Galdame, O.; Reggiardo, M.V.; Muñoz, A.E.; Adrover, R.; Cocozzella, D.; Fernandez, N.; Estepo, C.; Mendizábal, M.; et al. Relapse rates in chronic hepatitis B naive patients after discontinuation of antiviral therapy with entecavir. J. Viral Hepat. 2014, 21, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Wang, J.; Chen, X.; Xu, D.; Xia, N. Potential use of serum HBV RNA in antiviral therapy for chronic hepatitis B in the era of nucleos(t)ide analogs. Front. Med. 2017, 11, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Jung, H.Y.; Lee, K.H.; Yi, N.-J.; Suh, K.-S.; Jang, J.-J.; Lee, K.-B. Nuclear expression of hepatitis B virus X protein is associated with recurrence of early-stage hepatocellular carcinomas: Role of viral protein in tumor recurrence. J. Pathol. Transl. Med. 2016, 50, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Na, T.-Y.; Ka, N.L.; Rhee, H.; Kyeong, D.; Kim, M.H.; Seong, J.K.; Park, Y.N.; Lee, M.O. Interaction of hepatitis B virus X protein with PARP1 results in inhibition of DNA repair in hepatocellular carcinoma. Oncogene 2016, 35, 5435–5445. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Wakai, T.; Kubota, M.; Osawa, M.; Takamura, M.; Yamagiwa, S.; Ayoyagi, Y.; Sanpei, A.; Fujimaki, S. DNA damage sensor γ-H2AX is increased in preneoplastic lesions of hepatocellular carcinoma. Sci. World J. 2013, 2013, 597095. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, H.S.; Ji, J.H.; Cho, M.Y.; Yoo, Y.S.; Park, Y.Y.; Cha, H.J.; Lee, Y.; Kim, Y.; Cho, H. Hepatitis B virus X protein activates the ATM-Chk2 pathway and delays cell cycle progression. J. Gen. Virol. 2015, 96, 2242–2251. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Zhang, Y.; Cheng, L.; Ma, J.; Zi, Y.; Yang, L.; Su, C.; Shao, B.; Huang, A.; Xiang, R.; et al. Hepatitis B virus X protein (HBx)-induced abnormalities of nucleic acid metabolism revealed by 1H-NMR-based metabonomics. Sci. Rep. 2016, 6, 24430. [Google Scholar] [CrossRef]
- Murakami, Y.; Saigo, K.; Takashima, H.; Minami, M.; Okanoue, T.; Bréchot, C.; Paterlini-Bréchot, P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut 2005, 54, 1162–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadziyannis, S.J.; Papatheodoridis, G.V. Adefovir dipivoxil in the treatment of chronic hepatitis B virus infection. Expert Rev. Anti. Infect. Ther. 2004, 2, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Pol, S.; Lampertico, P. First-line treatment of chronic hepatitis B with entecavir or tenofovir in ‘real-life’ settings: From clinical trials to clinical practice. J. Viral Hepat. 2012, 19, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.F. Lamivudine monotherapy for chronic hepatitis B virus infection. Eur. J. Gastroenterol. Hepatol. 2009, 4, 447–451. [Google Scholar]
- Hartwell, D.; Jones, J.; Harris, P.; Cooper, K. Telbivudine for the treatment of chronic hepatitis B infection. Health Technol. Assess. 2009, 13, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Vigano, M.; Loglio, A.; Grossi, G.; Lampertico, P. Tenofovir alafenamide (TAF) treatment of HBV, what are the unanswered questions? Expert Rev. Anti. Infect. Ther. 2018, 16, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Konerman, M.A.; Lok, A.S. Interferon treatment for hepatitis B. Clin. Liver Dis. 2016, 20, 645–665. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Chung, Y.H.; Kim, J.A.; Jin, Y.J.; Park, W.H.; Kim, S.E.; Lee, D.; Shim, J.H.; Kim, K.M.; Lim, Y.S.; et al. rtL180M mutation of hepatitis B virus is closely associated with frequent virological resistance to adefovir dipivoxil therapy. J. Gastroenterol. Hepatol. 2012, 27, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Schildgen, O.; Sirma, H.; Funk, A.; Olotu, C.; Wend, U.C.; Hartmann, H.; Helm, M.; Rochstroh, J.K.; Willems, W.R.; Will, H.; et al. Variant of hepatitis B virus with primary resistance to adefovir. N. Engl. J. Med. 2006, 354, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-T.; Lai, C.L.; Kew Yoon, S.; Lee, S.S.; Coelho, H.S.; Carrolho, F.J.; Poordad, F.; Halota, W.; Horsmans, Y.; Tsai, N.; et al. Entecavir treatment for up to 5 years in patients with hepatitis B e antigen-positive chronic hepatitis B. Hepatology 2010, 51, 422–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenney, D.J.; Rose, R.E.; Baldick, C.J.; Pokornowski, K.A.; Eggers, B.J.; Fang, J.; Wichroski, M.J.; Xu, D.; Yang, J.; Wilber, R.B.; et al. Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. Hepatology 2009, 49, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Geipel, A.; Seiz, P.L.; Niekamp, H.; Neumann-Fraune, M.; Zhang, K.; Kaiser, R.; Protzer, U.; Gerlich, W.H.; Glebe, D.; HOPE Consortium. Entecavir allows an unexpectedly high residual replication of HBV mutants resistant to lamivudine. Antivir. Ther. 2015, 20, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Zoulim, F.; Hezode, C.; Causse, X.; Roche, B.; Truchi, R.; Pauwels, A.; Ouzan, D.; Dumortier, J.; Pageaux, G.P.; et al. Effectiveness and safety of tenofovir disoproxil fumarate in chronic hepatitis B: A 3-year, prospective, real-world study in France. Dig. Dis. Sci. 2016, 61, 3072–3083. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Hughes, S.L.; Gotham, D.; Pozniak, A.L. Tenofovir alafenamide versus tenofovir disoproxil fumarate: Is there a true difference in efficacy and safety? J. Virus Erad. 2018, 4, 72–79. [Google Scholar] [PubMed]
- Chan, H.L.; Fung, S.; Seto, W.K.; Gane, E.; Flaherty, J.F.; Suri, V.; Lin, L.; Gaggar, A.; Subramanian, G.M.; Chuang, W.L.; et al. Improved bone and renal safety of switching from tenofovir disoproxil fumarate to tenofovir alafenamide: Preliminary results from 2 phase 3 studies in HBeAg-positive and HBeAg-negative patients with chronic hepatitis B. J. Hepatol. 2017, 66, S25. [Google Scholar] [CrossRef]
- Buti, M.; Gane, E.; Seto, W.K.; Chan, H.L.Y.; Chuang, W.-L.; Stepanova, T.; Hui, A.J.; Lim, Y.-S.; Mehta, R.; Janssen, H.L.A.; et al. A phase 3 study of tenofovir alafenamide compared with tenofovir disoproxil fumarate in patients with HBeAg-negative, chronic hepatitis B: Week 48 efficacy and safety results. J. Hepatol. 2016, 64, S135–S136. [Google Scholar] [CrossRef]
- Schiff, E.R.; Lee, S.S.; Chao, Y.C.; Kew Yoon, S.; Bessone, F.; Wu, S.S.; Kryczka, W.; Lurie, Y.; Gadano, A.; Kitis, G.; et al. Long-term treatment with entecavir induces reversal of advanced fibrosis or cirrhosis in patients with chronic hepatitis B. Clin. Gastroenterol. Hepatol. 2011, 9, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Aguilar Schall, R.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- Lampertico, P.; Invernizzi, F.; Vigano, M.; Loglio, A.; Mangia, G.; Facchetti, F.; Primignani, M.; Jovani, M.; Lavarone, M.; Fraquelli, M.; et al. The long-term benefits of nucleos(t)ide analogs in compensated HBV cirrhotic patients with no or small esophageal varices: A 12-year prospective cohort study. J. Hepatol. 2015, 63, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Lampertico, P.; Soffredini, R.; Yurdaydin, C.; Idilman, R.; Papatheodoridis, G.V.; Margariti, A.; Buti, M.; Esteban, R.; Zaltron, S.; Vavassori, A.; et al. Four years of tenofovir monotherapy for NUC naïve field practice european patients suppresses HBV Replication in most patients with a favorable renal safety profile but does not prevent HCC in patients with or without cirrhosis. Dig. Liver Dis. 2014, 46, e14. [Google Scholar] [CrossRef]
- Papatheodoridis, G.; Dalekos, G.; Sypsa, V.; Yurdaydin, C.; Buti, M.; Goulis, J.; Calleja, J.L.; Chi, H.; Manolakopoulos, S.; Mangia, G.; et al. PAGE-B predicts the risk of developing hepatocellular carcinoma in caucasians with chronic hepatitis B on 5-year antiviral therapy. J. Hepatol. 2016, 64, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Janssen, N.L.A.; van Zonneveld, M.; Senturk, H.; Zeuzem, S.; Akarca, U.S.; Cakaloglu, Y.; Simon, C.; So, T.M.; Gerken, G.; de Man, R.A.; et al. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: A randomised trial. Lancet 2005, 365, 123–129. [Google Scholar] [CrossRef]
- Yapali, S.; Talaat, N.; Lok, A.S. Management of hepatitis B: Our practice and how it relates to the guidelines. Clin. Gastroenterol. Hepatol. 2014, 12, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Kartal, E.D.; Alpat, S.N.; Ozgunes, I.; Usluer, G. Adverse effects of high-dose interferon-alpha-2a treatment for chronic hepatitis B. Adv. Ther. 2007, 24, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Perrillo, R. Benefits and risks of interferon therapy for hepatitis B. Hepatology 2009, 49, S103–S111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Sohn, J.A.; Seeger, C. Distribution of hepatitis B virus nuclear DNA. J. Virol. 2018, 92, e01391-17. [Google Scholar] [CrossRef] [PubMed]
- Rivino, L.; Le Bert, N.; Gill, U.S.; Kunasegaran, K.; Cheng, Y.; Tan, D.Z.; Becht, E.; Hansi, N.K.; Foster, G.R.; Su, T.H.; et al. Hepatitis B virus-specific T cells associate with viral control upon nucleos(t)ide-analogue therapy discontinuation. J. Clin. Investig. 2018, 128, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Karimova, M.; Beschorner, N.; Dammermann, W.; Chemnitz, J.; Indenbirken, D.; Bockmann, J.H.; Grundhoff, A.; Lüth, S.; Buchholz, F.; Schulze zur Weish, J.; et al. CRISPR/Cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci. Rep. 2015, 5, 13734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, C.; Sohn, J.A. Targeting hepatitis B virus with CRISPR/Cas9. Mol. Ther. Nucleic Acids 2014, 3, e216. [Google Scholar] [CrossRef] [PubMed]
- Laras, A.; Koskinas, J.; Dimou, E.; Kostamena, A.; Hadziyannis, S.J. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology 2006, 44, 694–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-C.; Kao, J.-H. Persistence of hepatitis B virus covalently closed circular DNA in hepatocytes: Molecular mechanisms and clinical significance. Emerg. Microbes Infect. 2014, 3, e64. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sze, J.; He, M.-L. HBV cccDNA in patients’ sera as an indicator for HBV reactivation and an early signal of liver damage. World J. Gastroenterol. 2004, 10, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Werle-Lapostolle, B.; Bowden, S.; Locarnini, S.; Wursthorn, K.; Petersen, J.; Lau, G.; Trepo, C.; Marcellin, P.; Goodman, Z.; Delaney, W.E., 4th; et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy 1. Gastroenterology 2004, 126, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Takkenberg, B.; Terpstra, V.; Zaaijer, H.; Weegink, C.; Dijkgraaf, M.; Jansen, P.; Beld, M.; Reesink, H. Intrahepatic response markers in chronic hepatitis B patients treated with peginterferon alpha-2a and adefovir. J. Gastroenterol. Hepatol. 2011, 26, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, M.J.; Hanse, B.E.; Piratvisuth, T.; Jia, J.D.; Zeuzem, S.; Gane, E.; Liaw, Y.F.; Xie, Q.; Heathcote, E.J.; Chan, H.L.; et al. Response-guided peginterferon therapy in hepatitis B e antigen-positive chronic hepatitis B using serum hepatitis B surface antigen levels. Hepatology 2013, 58, 872–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuaypen, N.; Sriprapu, M.; Praianantathavorn, K.; Payungporn, S.; Wisedopas, N.; Poovorawan, Y.; Tangkijvanich, P. Kinetics of serum HBsAg and intrahepatic cccDNA during pegylated interferon therapy in patients with HBeAg-positive and HBeAg-negative chronic hepatitis B. J. Med. Virol. 2017, 89, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Puro, R.; Schneider, R.J. Tumor necrosis factor activates a conserved innate antiviral response to hepatitis B virus that destabilizes nucleocapsids and reduces nuclear viral DNA. J. Virol. 2007, 81, 7351–7362. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.E.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–E5724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mao, R.; Guo, H.; Zhang, J. Detection of HBV cccDNA methylation from clinical samples by bisulfite sequencing and methylation-specific PCR. Methods Mol. Biol. 2017, 1540, 73–84. [Google Scholar] [PubMed]
- Zhang, Y.; Mao, R.; Yan, R.; Cai, D.; Zhang, Y.; Zhu, H.; Kang, Y.; Liu, H.; Wang, J.; Qin, Y.; et al. Transcription of hepatitis B virus covalently closed circular DNA is regulated by CpG methylation during chronic infection. PLoS ONE 2014, 9, e110442. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Chang, T.-T.; Chen, S.; Boldbaatar, B.; Clemens, A.; Lin, S.Y.; Yan, R.; Hu, C.-T.; Guo, H.; Timothy, M.; et al. Comprehensive DNA methylation analysis of hepatitis B virus genome in infected liver tissues. Sci. Rep. 2015, 5, 10478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Li, Y.; Mu, S.; Zhang, J.; Yan, Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J. Med. Virol. 2009, 81, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Kostyushev, D.S.; Zueva, A.P.; Brezgin, S.A.; Lipatnikov, A.D.; Simirskii, V.N.; Glebe, D.; Volchkova, E.V; Shipulin, G.A.; Chulanov, V.P. Overexpression of DNA-methyltransferases in persistency of cccDNA pool in chronic hepatitis B. Ter. Arkh. 2017, 89, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kanai, Y.; Sakamoto, M.; Saito, H.; Ishii, H.; Hirohashi, S. Expression of mRNA for DNA methyltransferases and methyl-CpG–binding proteins and DNA methylation status on CpG islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology 2001, 33, 561–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivekanandan, P.; Thomas, D.; Torbenson, M. Hepatitis B viral DNA is methylated in liver tissues. J. Viral Hepat. 2008, 15, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, P.; Thomas, D.; Torbenson, M. Methylation regulates hepatitis B viral protein expression. J. Infect. Dis. 2009, 199, 1286–1291. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, P.; Daniel, H.D.-J.; Kannangai, R.; Martinez-Murillo, F.; Torbenson, M. Hepatitis B virus replication induces methylation of both host and viral DNA. J. Virol. 2010, 84, 4321–4329. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Paliwal, A.; Durantel, D.; Hainaut, P.; Scoazec, J.Y.; Zoulim, F.; Chemin, I.; Herceg, Z. DNA methylation of hepatitis B virus (HBV) genome associated with the development of hepatocellular carcinoma and occult HBV infection. J. Infect. Dis. 2010, 202, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Allday, M.J. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol. 2010, 18, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köck, J.; Theilmann, L.; Galle, P.; Schlicht, H.J. Hepatitis B virus nucleic acids associated with human peripheral blood mononuclear cells do not originate from replicating virus. Hepatology 1996, 23, 405–413. [Google Scholar] [PubMed] [Green Version]
- Luckenbaugh, L.; Kitrinos, K.M.; Delaney, W.E., 4th; Hu, J. Genome-free hepatitis B virion levels in patient sera as a potential marker to monitor response to antiviral therapy. J. Viral Hepat. 2015, 22, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Nguyen, D.; Mentzer, L.; Adams, C.; Lee, H.; Ashley, R.; Hafenstein, S.; Hu, J. Secretion of genome-free hepatitis B virus-single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog. 2011, 7, e1002255. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, J.; Li, W.; Chen, R.; Chen, X.; Zhang, F.; Xu, D.; Lu, F. Serum HBV DNA plus RNA shows superiority in reflecting the activity of intrahepatic cccDNA in treatment-naive HBV-infected individuals. J. Clin. Virol. 2018, 99–100, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Van Campenhout, M.J.H.; van Bömmel, F.; Pfefferkorn, M.; Fischer, J.; Deichsel, D.; Boonstra, A.; van Vuuren, A.J.; Berg, T.; Hansen, B.E.; Janssen, H.L.A. Host and viral factors associated with serum hepatitis B virus RNA levels among patients in need for treatment. Hepatology 2018, 28, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Jansen, L.; Kootstra, N.A.; van Dort, K.A.; Takkenberg, R.B.; Reesink, H.W.; Zaaijer, H.L. Hepatitis B virus pregenomic RNA is present in virions in plasma and is associated with a response to pegylated interferon alfa-2a and nucleos(t)ide analogues. J. Infect. Dis. 2016, 213, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-W.; Huang, Y.W.; Takahashi, S.; Tsuge, M.; Chen, C.L.; Wang, T.C.; Abe, H.; Hu, J.T.; Chen, D.S.; Yang, S.S.; et al. On-treatment low serum HBV RNA level predicts initial virological response in chronic hepatitis B patients receiving nucleoside analogue therapy. Antivir. Ther. 2015, 20, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, M.; Murakami, E.; Imamura, M.; Abe, H.; Miki, D.; Hiraga, N.; Takahashi, S.; Ochi, H.; Nelson Hayes, C.; Ginba, H.; et al. Serum HBV RNA and HBeAg are useful markers for the safe discontinuation of nucleotide analogue treatments in chronic hepatitis B patients. J. Gastroenterol. 2013, 48, 1188–1204. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, F.; Wang, L.; Liu, Y.; Zhang, M.; Li, T. Clinical characteristics and outcomes of patients with recurrent chronic hepatitis B after nucleos(t)ide analog withdrawal with stringent cessation criteria: A prospective cohort study. Hepatol. Res. 2017, 47, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Cornberg, M.; Wong, V.W.-S.; Locarnini, S.; Brunetto, M.; Janssen, H.L.A.; Chan, H.L.-Y. The role of quantitative hepatitis B surface antigen revisited. J. Hepatol. 2017, 66, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.L.; Chung, R.T. Monitoring during and after antiviral therapy for hepatitis B. Hepatology 2009, 49, S166–S173. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Yang, S.S.; Su, C.W.; Wang, Y.J.; Lee, K.C.; Huo, T.I.; Lin, H.C.; Huang, Y.H. Predictors of response to pegylated interferon in chronic hepatitis B: A real-world hospital-based analysis. Sci. Rep. 2016, 6, 29605. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Perrillo, R.P. Serum alanine aminotransferase flares during interferon treatment of chronic hepatitis B: Is sustained clearance of HBV DNA dependent on levels of pretreatment viremia? Hepatology 2001, 34, 1021–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farag, M.; Campenhout, M.V.; van Bömmel, F.; Rijckborst, V.; Cakaloglu, Y.; Ferenci, P.; Tabak, F.; Feld, F.; Hansen, B.; Janssen, H. HBV RNA in serum is an early predictor for sustained immune control following treatment with pegylated interferon alfa in patients with HBeAg-negative chronic hepatitis B. J. Hepatol. 2018, 68, S511. [Google Scholar] [CrossRef]
- Moore, M.J.; Silver, P.A. Global analysis of mRNA splicing. RNA 2008, 14, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Magen, A.; Ast, G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2017, 35, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.-N.; Chong, C.L.; Chou, Y.C.; Huang, C.C.; Wang, Y.L.; Wang, S.W.; Chen, M.L.; Chen, C.H.; Chang, C. Doubly spliced RNA of hepatitis B virus suppresses viral transcription via TATA-binding protein and induces stress granule assembly. J. Virol. 2015, 89, 11406–11419. [Google Scholar] [CrossRef] [PubMed]
- Candotti, D.; Allain, J.-P. Biological and clinical significance of hepatitis B virus RNA splicing: An update. Ann. Blood. 2017, 2, 1–14. [Google Scholar] [CrossRef]
- Wu, H.L.; Chen, P.J.; Tu, S.J.; Lin, M.H.; Lai, M.Y.; Chen, D.S. Characterization and genetic analysis of alternatively spliced transcripts of hepatitis B virus in infected human liver tissues and transfected HepG2 cells. J. Virol. 1991, 65, 1680–1686. [Google Scholar] [PubMed]
- Soussan, P.; Tuveri, R.; Nalpas, B.; Garreau, F.; Zavala, F.; Masson, A.; Pol, S.; Brechot, C.; Kremsdorf, D. The expression of hepatitis B spliced protein (HBSP) encoded by a spliced hepatitis B virus RNA is associated with viral replication and liver fibrosis. J. Hepatol. 2003, 38, 343–348. [Google Scholar] [CrossRef]
- Pol, J.G.; Lekbaby, B.; Redelsperger, F.; Klamer, S.; Mandouri, Y.; Ahodantin, J.; Bieche, I.; Lefevre, M.; Souque, P.; Charneau, P.; et al. Alternative splicing-regulated protein of hepatitis B virus hacks the TNF-alpha-stimulated signaling pathways and limits the extent of liver inflammation. FASEB J. 2015, 29, 1879–1889. [Google Scholar] [CrossRef] [PubMed]
- Duriez, M.; Mandouri, Y.; Lekbaby, B.; Wang, H.; Schnuriger, A.; Redelsperger, F.; Guerrera, C.I.; Lefevre, M.; Fauveau, V.; Ahodantin, J.; et al. Alternative splicing of hepatitis B virus: A novel virus/host interaction altering liver immunity. J. Hepatol. 2017, 67, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Soussan, P.; Pol, J.; Garreau, F.; Schneider, V.; Le Pendeven, C.; Nalpas, B.; Lacombe, K.; Bonnard, P.; Pol, S.; Kremsdorf, D. Expression of defective hepatitis B virus particles derived from singly spliced RNA is related to liver disease. J. Infect. Dis. 2008, 198, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Preiss, S.; Littlejohn, M.; Angus, P.; Thompson, A.; Desmond, P.; Lewin, S.R.; Sasadeusz, J.; Matthews, G.; Dore, G.S.; Shaw, T.; et al. Defective hepatitis B virus DNA is not associated with disease status but is reduced by polymerase mutations associated with drug resistance. Hepatology 2008, 48, 741–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.-M.; Lin, X.; Wang, Y.-X.; Tian, X.-C.; Xie, Y.-H.; Wen, Y.-M. A double-spliced defective hepatitis B virus genome derived from hepatocellular carcinoma tissue enhanced replication of full-length virus. J. Med. Virol. 2009, 81, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-T.; Wong, J.K.; Choi, S.W.; Sze, K.M.; Ho, D.W.; Chan, L.K.; Lee, J.M.; Man, K.; Cherny, S.; Yang, W.L.; et al. Novel pre-mRNA splicing of intronically integrated HBV generates oncogenic chimera in hepatocellular carcinoma. J. Hepatol. 2016, 64, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, A.; Blondin, D.; Hauck, K.; Sagir, A.; Kohnle, T.; Heintges, T.; Häussinger, D. Response to interferon alfa is hepatitis B virus genotype dependent: Genotype A is more sensitive to interferon than genotype D. Gut 2005, 54, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Nie, H.; Yan, R.; Guo, J.-T.; Block, T.M.; Guo, H. A southern blot assay for detection of hepatitis B virus covalently closed circular DNA from cell cultures. Methods Mol. Biol. 2013, 1030, 151–161. [Google Scholar] [PubMed]
- Wong, D.K.-H.; Yuen, M.F.; Yuan, H.; Sum, S.S.; Hui, C.K.; Hall, J.; Lai, C.L. Quantitation of covalently closed circular hepatitis B virus DNA in chronic hepatitis B patients. Hepatology 2004, 40, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addison, W.R.; Wong, W.W.; Fischer, K.P.; Tyrrell, D.L. A quantitative competitive PCR assay for the covalently closed circular form of the duck hepatitis B virus. Antiviral Res. 2000, 48, 27–37. [Google Scholar] [CrossRef]
- Arad, U. Modified Hirt procedure for rapid purification of extrachromosomal DNA from mammalian cells. Biotechniques 1998, 24, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Rybicka, M.; Woziwodzka, A.; Romanowski, T.; Stalke, P.; Dręczewski, M.; Bielawski, K.P. Differences in sequences between HBV-relaxed circular DNA and covalently closed circular DNA. Emerg. Microbes Infect. 2017, 6, e55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.-T.; Han, T.; Li, Y.; Yang, B.; Wang, Y.J.; Wang, F.M.; Jing, X.; Du, Z. Enhanced specificity of real-time PCR for measurement of hepatitis B virus cccDNA using restriction endonuclease and plasmid-safe ATP-dependent DNase and selective primers. J. Virol. Methods 2010, 169, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Sheng, S.; Nie, B.; Tu, Z. Development of magnetic capture hybridization and quantitative polymerase chain reaction for hepatitis B virus covalently closed circular DNA. Hepat. Mon. 2015, 15, e23729. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-T.; Yang, Y.; Hu, Y.M.; Liu, X.H.; Liao, M.Y.; Morgan, R.; Yuan, E.F.; Li, X.; Liu, S.M. A highly sensitive and robust method for hepatitis B virus covalently closed circular DNA detection in single cells and serum. J. Mol. Diagn. 2018, 20, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; McConkey, M.E.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Nelles, D.A.; Fang, M.Y.; O’Connell, M.R.; Xu, J.L.; Doudna, J.A.; Yeo, G.W. Programmable RNA tracking in live cells with CRISPR/Cas9. Cell 2016, 165, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Naseri, A.; Reyes-Gutierrez, P.; Wolfe, S.A.; Zhang, S.; Pederson, T. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. USA 2015, 112, 3002–3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, D.K.-H.; Seto, W.K.; Cheung, K.S.; Chong, C.K.; Huang, F.Y.; Fug, J.; Lai, C.L.; Yuen, M.F. Hepatitis B virus core-related antigen as a surrogate marker for covalently closed circular DNA. Liver Int. 2017, 37, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Rampazzo, R.D.C.P.; Costa, A.D.T.; Krieger, M.A. Current nucleic acid extraction methods and their implications to point-of-care diagnostics. BioMed Res. Int. 2017, 9306564. [Google Scholar] [CrossRef] [PubMed]
- Butler, E.K.; Gersch, J.; Luk, K.C.; Holzmayer, V.; de Medina, M.; Schiff, E.; Kuhns, M.; Cloherty, G.A. Hepatitis B virus serum DNA and RNA levels in nucleos(t)ide analog-treated or untreated patients during chronic and acute infection. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Therapeutic Agents | Representative Drugs | Phase of Clinical Trials |
|---|---|---|
| Inhibition of HBV entry | Myrcludex-B | Phase II clinical trials |
| Degradation of cccDNA | CRISPR/Cas9, APOBEC-deaminases, LT-βR agonist | Preclinical studies |
| Capsid assembly inhibitors | GLS4, NVR 3-778, AIC 649, ABI-H0731 | Phases I-II clinical trials |
| miRNA | ARB-1467, ARB-1740 | Phase II clinical trials |
| Therapeutic vaccinations | INO-1800, HB-110, TG1050, HepTcell | Phase I clinical trials |
| Intracellular immune response agonists | GS 9620, SB9200, AIC649 | Phase II clinical trials |
| cccDNA inhibitors | CCC-0975, CCC-0346 | Preclinical studies |
| HBsAg inhibitors | Rep 2139, Rep 2055 | II phase of clinical trials |
| Marker | Applications | Result |
|---|---|---|
| cccDNA | Defining absolute cure | cccDNA is undetectable. |
| Predicting IFN responsiveness in HBeAg-positive patients | cccDNA level is lower in IFN responders than non-responders. | |
| Serum HBV RNA | Safely discontinuing NA therapy | HBV RNA is undetectable. |
| Predicting YMDD mutations | High serum HBV RNA levels predict lamivudine resistance after the first year of treatment. | |
| Predicting HBeAg seroconversion in HBeAg-positive patients receiving IFN | HBV RNA levels > 5.5; log10 copies/mL predict non-responders to IFN therapy (weeks 12 and 24). | |
| Predicting IFN responsiveness in HBeAg-negative patients. | High levels of serum HBV RNA are a reliable marker of non-responsiveness to IFN therapy (week 12). | |
| HBV RNA splice variants | Predicting IFN responsiveness | Elevated HBV splice variants in serum negatively correlate with responsiveness to IFN treatment; HBV DNA splice variants in the serum reflect the levels of intracellular HBV RNA splice variants. |
| Factors | Effect |
|---|---|
| Presence of BCP variants | Lower HBV RNA serum levels |
| HBV genotype | Patients with HBV of genotypes A, B, and C have lower HBV RNA serum levels than of genotype D |
| ALT levels | Higher in patients with ALT level > 2 × upper limit of normal (ULN) compared to patients with ALT level < 2 × ULN |
| Patient’s age | No influence |
| Patient’s sex | No influence |
| Parameter | HBeAg-Positive | HBeAg-Negative |
|---|---|---|
| Mean serum HBV RNA level | 6.5 (1.2) log c/mL | 4.1 (1.2) log c/mL |
| Correlation of serum HBV RNA and HBV DNA | Strong | Strong |
| Correlation between HBV RNA and HBsAg | Moderate | Weak |
| Method | Specificity | Limit of Detection | Advantages | Disadvantages | |
|---|---|---|---|---|---|
| cccDNA | Southern blotting | Unequivocally determines cccDNA | 2 × 106 copies | Reliable; reproducible | Complicated; costly; time-consuming; safety concerns |
| Conventional qPCR | May under- or overrepresent cccDNA | 2 × 103 copies/mL | Simple; rapid; accurate; economical, sensitive | Lower specificity when rcDNA is abundant | |
| Competitive qPCR | More specific than conventional qPCR; may still overrepresent cccDNA by amplifying rcDNA | 2 × 104 copies | More specific and accurate than conventional qPCR; readily distinguishes cccDNA from rcDNA | Lower specificity when rcDNA is abundant | |
| Droplet-digital PCR | Specific | 1 copy; upper detection limit is restricted | Super-sensitive; accurate | Detection is impaired when cccDNA number is greater than 106 copies | |
| Rolling circle amplification qPCR | Specific | 102 copies/mL | Practical; sensitive; specific | Time-consuming; cross-linked proteins impair effective amplification | |
| Rolling circle amplification-in situ qPCR | Highly specific; cross-linked proteins could hinder effective amplification | 2 copies/cell | cccDNA detection at single-cell resolution | Diffusion of amplified DNA to neighboring cells; cross-linked proteins impair effective amplification | |
| Magnetic capture hybridization qPCR | Specific | 90 IU/mL | Specific | Does not capture all cccDNA; complicated; costly | |
| Invader assay | Specific; minimal interference from double-stranded and integrated HBV DNA | 50 copies (104 copies/mL) | Provides a specific and simple method for detecting cccDNA comparable with PCR | Interference from rcDNA and integrated HBV DNA | |
| FISH detection | Specific; distinguishes cccDNA at single-cell resolution; no diffusion of amplified products | 1 copy under optimal conditions | Specific; visible at single-cell resolution; can distinguish and locate various DNA, RNA and proteins; without diffusion of amplified products | Complicated probe design | |
| Semi-nested and nested qPCR | Specific | 3.0 × 102 copies/mL | May be contaminated by PCR products | ||
| HBV RNA | RUO HBV RNA assay (Abbot) | Highly specific | 44 IU/mL | Highly sensitive and specific; automated; high throughput | |
| RACE-based methods | Specific | 54 IU/mL | No additional steps in isolation procedure; sensitive; specific | ||
| Detection after DNase I treatment | Specific | 66.7 IU/mL | Requires complicated isolation procedure (DNase I treatment and purification); time-consuming; allows enrichment of RNA compared to isolated DNA (does not eliminate all DNA) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostyusheva, A.; Kostyushev, D.; Brezgin, S.; Volchkova, E.; Chulanov, V. Clinical Implications of Hepatitis B Virus RNA and Covalently Closed Circular DNA in Monitoring Patients with Chronic Hepatitis B Today with a Gaze into the Future: The Field Is Unprepared for a Sterilizing Cure. Genes 2018, 9, 483. https://doi.org/10.3390/genes9100483
Kostyusheva A, Kostyushev D, Brezgin S, Volchkova E, Chulanov V. Clinical Implications of Hepatitis B Virus RNA and Covalently Closed Circular DNA in Monitoring Patients with Chronic Hepatitis B Today with a Gaze into the Future: The Field Is Unprepared for a Sterilizing Cure. Genes. 2018; 9(10):483. https://doi.org/10.3390/genes9100483
Chicago/Turabian StyleKostyusheva, Anastasiya, Dmitry Kostyushev, Sergey Brezgin, Elena Volchkova, and Vladimir Chulanov. 2018. "Clinical Implications of Hepatitis B Virus RNA and Covalently Closed Circular DNA in Monitoring Patients with Chronic Hepatitis B Today with a Gaze into the Future: The Field Is Unprepared for a Sterilizing Cure" Genes 9, no. 10: 483. https://doi.org/10.3390/genes9100483
APA StyleKostyusheva, A., Kostyushev, D., Brezgin, S., Volchkova, E., & Chulanov, V. (2018). Clinical Implications of Hepatitis B Virus RNA and Covalently Closed Circular DNA in Monitoring Patients with Chronic Hepatitis B Today with a Gaze into the Future: The Field Is Unprepared for a Sterilizing Cure. Genes, 9(10), 483. https://doi.org/10.3390/genes9100483