DNA Damage Tolerance Mechanisms Revealed from the Analysis of Immunoglobulin V Gene Diversification in Avian DT40 Cells

Abstract
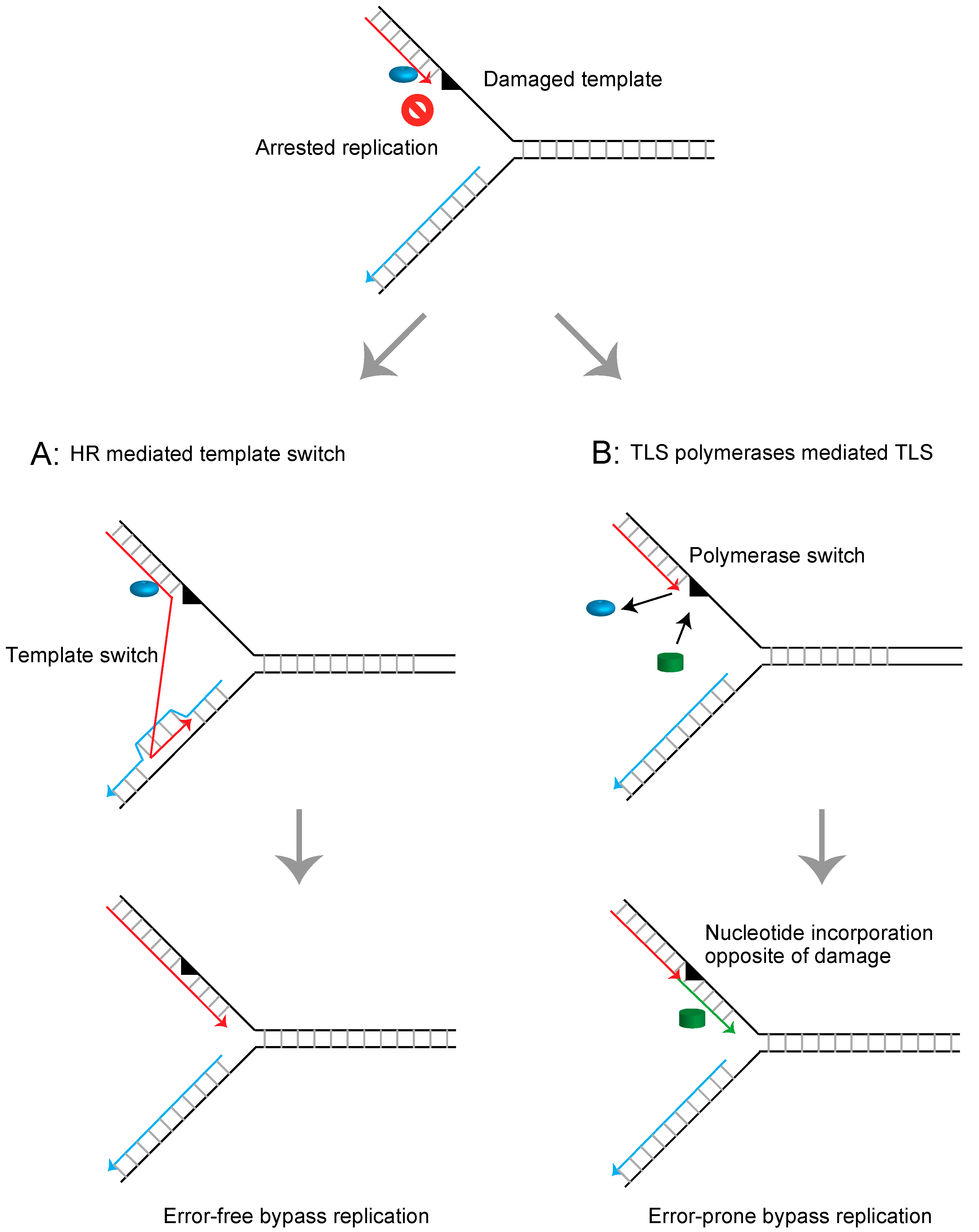
:1. Introduction
2. Immunoglobulin variable (IgV) Gene Diversification in DT40 Cells
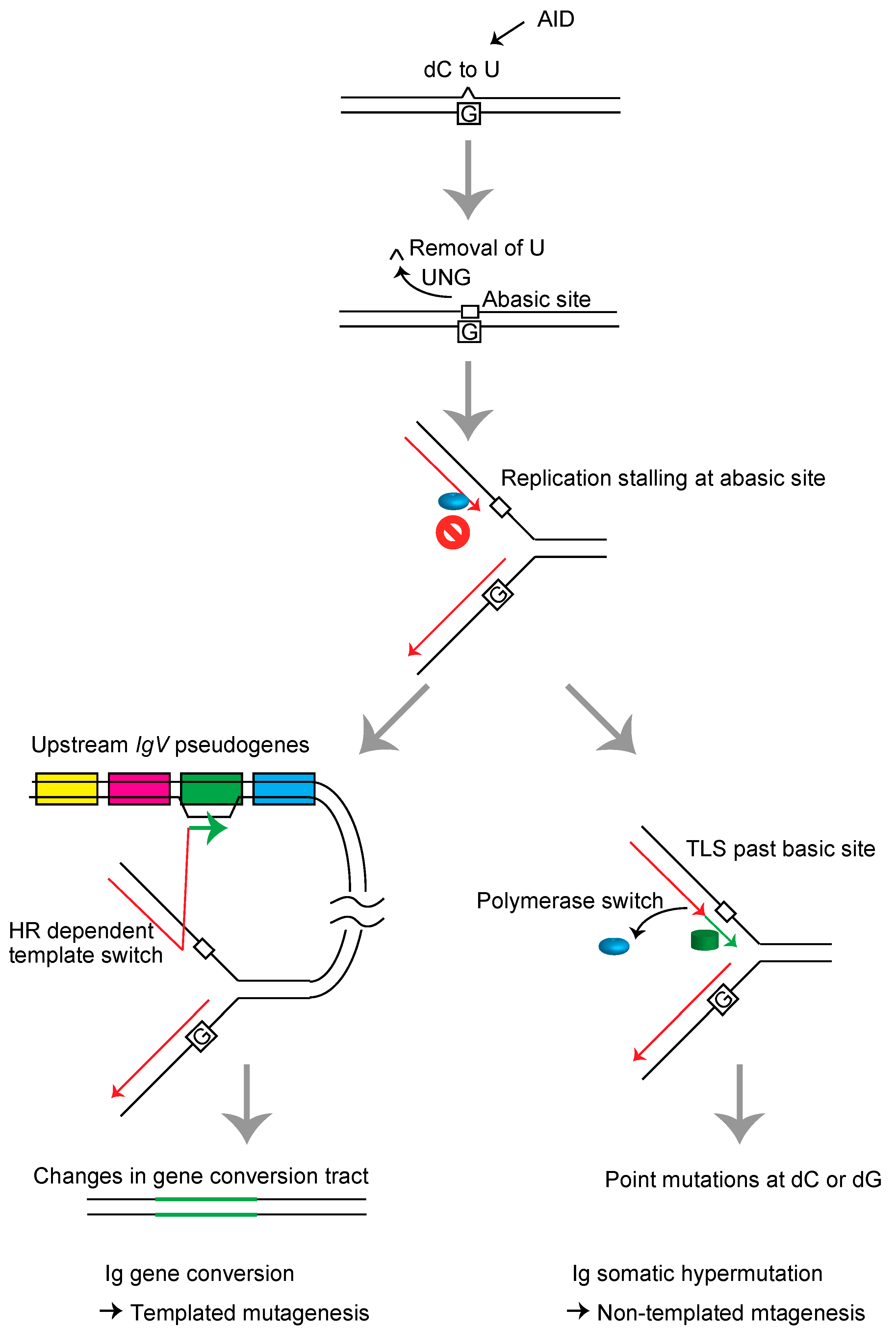
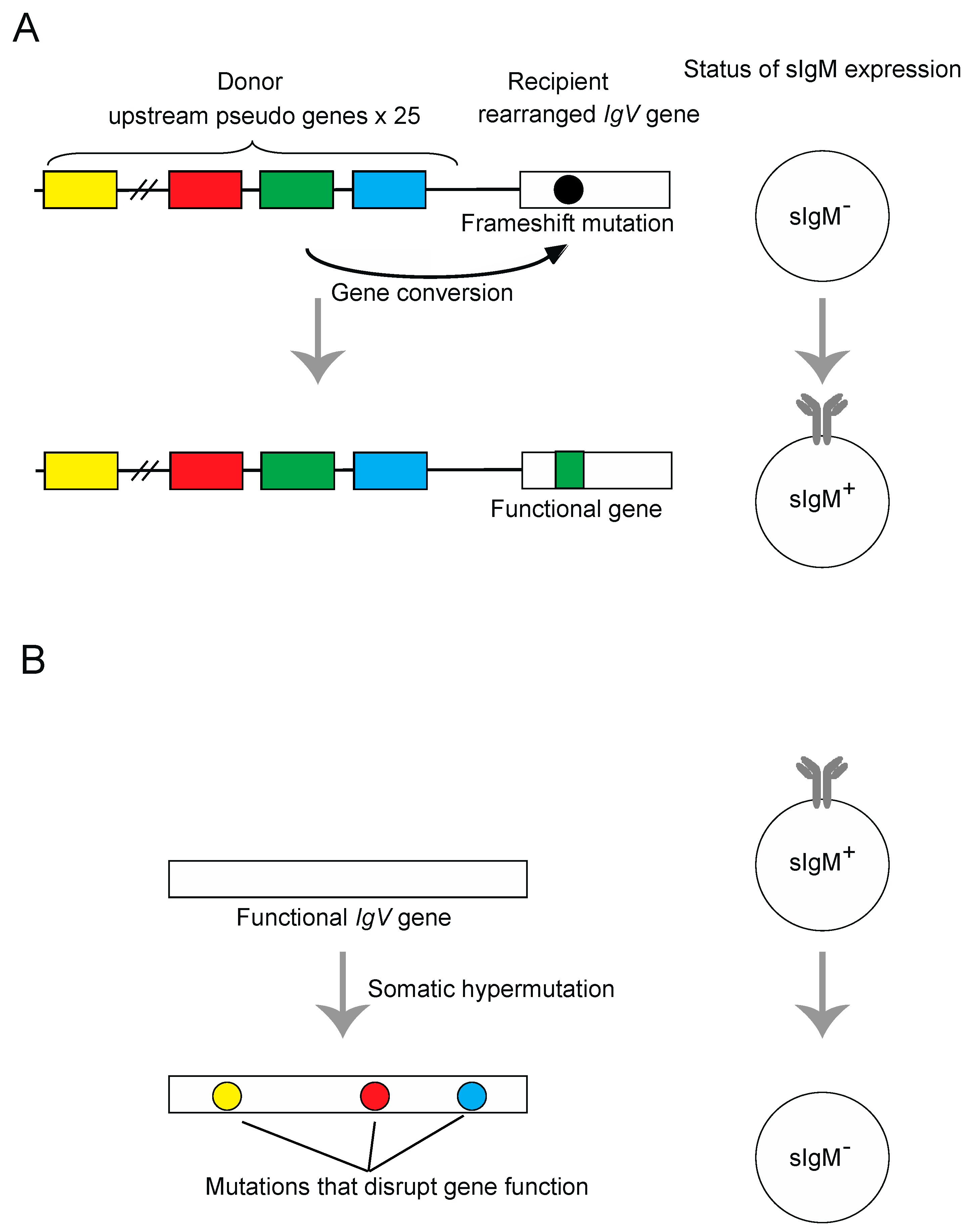
3. Templated Mutagenesis by Gene Conversion
4. Nontemplated Somatic Hypermutation
5. Summary and Perspective
Funding
Conflicts of Interest
References
- McCulloch, S.D.; Kunkel, T.A. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008, 18, 148–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, M.W.; Matsumoto, Y.; Loeb, L.A. High fidelity and lesion bypass capability of human DNA polymerase delta. Biochimie 2009, 91, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Boston, MA, USA, 2006. [Google Scholar]
- Barnes, D.E.; Lindahl, T.; Sedgwick, B. DNA repair. Curr. Opin. Cell Biol. 1993, 5, 424–433. [Google Scholar] [CrossRef]
- Klungland, A.; Rosewell, I.; Hollenbach, S.; Larsen, E.; Daly, G.; Epe, B.; Seeberg, E.; Lindahl, T.; Barnes, D.E. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13300–13305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeb, L.A. Apurinic sites as mutagenic intermediates. Cell 1985, 40, 483–484. [Google Scholar] [CrossRef]
- Prasad, R.; Horton, J.K.; Liu, Y.; Wilson, S.H. Central steps in mammalian BER and regulation by PARP1. Base Excision Repair Pathw. 2017, 253–280. [Google Scholar] [CrossRef]
- Branzei, D.; Psakhye, I. DNA damage tolerance. Curr. Opin. Cell Biol. 2016, 40, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Mutter-Rottmayer, E.; Zlatanou, A.; Vaziri, C.; Yang, Y. Mechanisms of post-replication DNA repair. Genes 2017, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Kasaishi, Y.; Nakada, S.; Takagi, T.; Era, S.; Motegi, A.; Chiu, R.K.; Takeda, S.; Hirota, K. Rad18 and Rnf8 facilitate homologous recombination by two distinct mechanisms, promoting Rad51 focus formation and suppressing the toxic effect of nonhomologous end joining. Oncogene 2015, 34, 4403–4411. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, K.; Sonoda, E.; Kawamoto, T.; Motegi, A.; Masutani, C.; Hanaoka, F.; Szuts, D.; Iwai, S.; Sale, J.E.; Lehmann, A.; et al. Simultaneous disruption of two DNA polymerases, Poleta and Polzeta, in Avian DT40 cells unmasks the role of Poleta in cellular response to various DNA lesions. PLoS Genet. 2010, 6, e1001151. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Tsuda, M.; Tsurimoto, T.; Cohen, I.S.; Livneh, Z.; Kobayashi, K.; Narita, T.; Nishihara, K.; Murai, J.; Iwai, S.; et al. In vivo evidence for translesion synthesis by the replicative DNA polymerase delta. Nucleic Acids Res. 2016, 44, 7242–7250. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Yoshikiyo, K.; Guilbaud, G.; Tsurimoto, T.; Murai, J.; Tsuda, M.; Phillips, L.G.; Narita, T.; Nishihara, K.; Kobayashi, K.; et al. The POLD3 subunit of DNA polymerase δ can promote translesion synthesis independently of DNA polymerase ζ. Nucleic Acids Res. 2015, 43, 1671–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohzaki, M.; Nishihara, K.; Hirota, K.; Sonoda, E.; Yoshimura, M.; Ekino, S.; Butler, J.E.; Watanabe, M.; Halazonetis, T.D.; Takeda, S. DNA polymerases ν and θ are required for efficient immunoglobulin V gene diversification in chicken. J. Cell Biol. 2010, 189, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Sale, J.E. Competition, collaboration and coordination–determining how cells bypass DNA damage. J. Cell Sci. 2012, 125, 1633–1643. [Google Scholar] [CrossRef]
- Arakawa, H.; Buerstedde, J.M. Activation-induced cytidine deaminase-mediated hypermutation in the DT40 cell line. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef]
- Sale, J.E.; Batters, C.; Edmunds, C.E.; Phillips, L.G.; Simpson, L.J.; Szuts, D. Timing matters: Error-prone gap filling and translesion synthesis in immunoglobulin gene hypermutation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 595–603. [Google Scholar] [CrossRef]
- Romanello, M.; Schiavone, D.; Frey, A.; Sale, J.E. Histone H3.3 promotes IgV gene diversification by enhancing formation of AID-accessible single-stranded DNA. EMBO J. 2016, 35, 1452–1464. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Felsenfeld, G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007, 21, 1519–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Noia, J.; Neuberger, M.S. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 2002, 419, 43–48. [Google Scholar] [CrossRef]
- Saribasak, H.; Saribasak, N.N.; Ipek, F.M.; Ellwart, J.W.; Arakawa, H.; Buerstedde, J.M. Uracil DNA glycosylase disruption blocks Ig gene conversion and induces transition mutations. J. Immunol. 2006, 176, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Blagodatski, A.; Batrak, V.; Schmidl, S.; Schoetz, U.; Caldwell, R.B.; Arakawa, H.; Buerstedde, J.M. A cis-acting diversification activator both necessary and sufficient for AID-mediated hypermutation. PLoS Genet. 2009, 5, e1000332. [Google Scholar] [CrossRef]
- Arakawa, H.; Moldovan, G.L.; Saribasak, H.; Saribasak, N.N.; Jentsch, S.; Buerstedde, J.M. A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 2006, 4, e366. [Google Scholar] [CrossRef]
- Arakawa, H.; Saribasak, H.; Buerstedde, J.M. Activation-induced cytidine deaminase initiates immunoglobulin gene conversion and hypermutation by a common intermediate. PLoS Biol. 2004, 2, e179. [Google Scholar] [CrossRef]
- Di Noia, J.M.; Neuberger, M.S. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22. [Google Scholar] [CrossRef]
- Sale, J.E. Immunoglobulin diversification in DT40: A model for vertebrate DNA damage tolerance. DNA Repair 2004, 3, 693–702. [Google Scholar] [CrossRef]
- Sale, J.E. Measurement of diversification in the immunoglobulin light chain gene of DT40 cells. Methods Mol. Biol. 2012, 920, 417–432. [Google Scholar] [CrossRef]
- Buerstedde, J.M.; Reynaud, C.A.; Humphries, E.H.; Olson, W.; Ewert, D.L.; Weill, J.C. Light chain gene conversion continues at high rate in an ALV-induced cell line. EMBO J. 1990, 9, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Buerstedde, J.M.; Takeda, S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell 1991, 67, 179–188. [Google Scholar] [CrossRef]
- Seo, H.; Masuoka, M.; Murofushi, H.; Takeda, S.; Shibata, T.; Ohta, K. Rapid generation of specific antibodies by enhanced homologous recombination. Nat. Biotechnol. 2005, 23, 731–735. [Google Scholar] [CrossRef]
- Cummings, W.J.; Yabuki, M.; Ordinario, E.C.; Bednarski, D.W.; Quay, S.; Maizels, N. Chromatin structure regulates gene conversion. PLoS Biol. 2007, 5, e246. [Google Scholar] [CrossRef]
- Arakawa, H. Immunoglobulin gene conversion and hypermutation assay by FACs. Sub-Cell. Biochem. 2006, 40, 351–352. [Google Scholar]
- Shinkura, R.; Ito, S.; Begum, N.A.; Nagaoka, H.; Muramatsu, M.; Kinoshita, K.; Sakakibara, Y.; Hijikata, H.; Honjo, T. Separate domains of AID are required for somatic hypermutation and class-switch recombination. Nat. Immunol. 2004, 5, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Saberi, A.; Nakahara, M.; Sale, J.E.; Kikuchi, K.; Arakawa, H.; Buerstedde, J.M.; Yamamoto, K.; Takeda, S.; Sonoda, E. The 9-1-1 DNA clamp is required for immunoglobulin gene conversion. Mol. Cell. Biol. 2008, 28, 6113–6122. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, Z.; Rada, C.; Neuberger, M.S. AID upmutants isolated using a high-throughput screen highlight the immunity/cancer balance limiting DNA deaminase activity. Nat. Struct. Mol. Biol. 2009, 16, 769–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sale, J.E.; Calandrini, D.M.; Takata, M.; Takeda, S.; Neuberger, M.S. Ablation of XRCC2/3 transforms immunoglobulin V gene conversion into somatic hypermutation. Nature 2001, 412, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.L.; Sale, J.E. The catalytic activity of REV1 is employed during immunoglobulin gene diversification in DT40. Mol. Immunol. 2006, 43, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Longerich, S.; Orelli, B.J.; Martin, R.W.; Bishop, D.K.; Storb, U. Brca1 in immunoglobulin gene conversion and somatic hypermutation. DNA Repair 2008, 7, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatanaka, A.; Yamazoe, M.; Sale, J.E.; Takata, M.; Yamamoto, K.; Kitao, H.; Sonoda, E.; Kikuchi, K.; Yonetani, Y.; Takeda, S. Similar effects of Brca2 truncation and Rad51 paralog deficiency on immunoglobulin V gene diversification in DT40 cells support an early role for Rad51 paralogs in homologous recombination. Mol. Cell. Biol. 2005, 25, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Bezzubova, O.; Silbergleit, A.; Yamaguchi-Iwai, Y.; Takeda, S.; Buerstedde, J.M. Reduced X-ray resistance and homologous recombination frequencies in a RAD54−/− mutant of the chicken DT40 cell line. Cell 1997, 89, 185–193. [Google Scholar] [CrossRef]
- Yamamoto, K.; Hirano, S.; Ishiai, M.; Morishima, K.; Kitao, H.; Namikoshi, K.; Kimura, M.; Matsushita, N.; Arakawa, H.; Buerstedde, J.M.; et al. Fanconi anemia protein FANCD2 promotes immunoglobulin gene conversion and DNA repair through a mechanism related to homologous recombination. Mol. Cell. Biol. 2005, 25, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, T.; Araki, K.; Sonoda, E.; Yamashita, Y.M.; Harada, K.; Kikuchi, K.; Masutani, C.; Hanaoka, F.; Nozaki, K.; Hashimoto, N.; et al. Dual roles for DNA polymerase eta in homologous DNA recombination and translesion DNA synthesis. Mol. Cell 2005, 20, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Holzschu, D.L.; Sugiyama, T. PCNA is efficiently loaded on the DNA recombination intermediate to modulate polymerase δ, η, and ζ activities. Proc. Natl. Acad. Sci. USA 2013, 110, 7672–7677. [Google Scholar] [CrossRef] [PubMed]
- McIlwraith, M.J.; Vaisman, A.; Liu, Y.; Fanning, E.; Woodgate, R.; West, S.C. Human DNA polymerase η promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol. Cell 2005, 20, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Sebesta, M.; Burkovics, P.; Haracska, L.; Krejci, L. Reconstitution of DNA repair synthesis in vitro and the role of polymerase and helicase activities. DNA Repair 2011, 10, 567–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebesta, M.; Burkovics, P.; Juhasz, S.; Zhang, S.; Szabo, J.E.; Lee, M.Y.; Haracska, L.; Krejci, L. Role of PCNA and TLS polymerases in D-loop extension during homologous recombination in humans. DNA Repair 2013, 12, 691–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase η with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D. Deubiquitinating PCNA: A downside to DNA damage tolerance. Nat. Cell Biol. 2006, 8, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Navadgi-Patil, V.M.; Burgers, P.M. The unstructured C-Terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol. Cell 2009, 36, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Navadgi-Patil, V.M.; Burgers, P.M. A tale of two tails: Activation of DNA damage checkpoint kinase Mec1/ATR by the 9-1-1 clamp and by Dpb11/TopBP1. DNA Repair 2009, 8, 996–1003. [Google Scholar] [CrossRef] [Green Version]
- Niida, H.; Nakanishi, M. DNA damage checkpoints in mammals. Mutagenesis 2006, 21, 3–9. [Google Scholar] [CrossRef]
- Bermudez, V.P.; Lindsey-Boltz, L.A.; Cesare, A.J.; Maniwa, Y.; Griffith, J.D.; Hurwitz, J.; Sancar, A. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 1633–1638. [Google Scholar] [CrossRef] [Green Version]
- Ellison, V.; Stillman, B. Biochemical characterization of DNA damage checkpoint complexes: Clamp loader and clamp complexes with specificity for 5’ recessed DNA. PLoS Biol. 2003, 1, E33. [Google Scholar] [CrossRef]
- Budzowska, M.; Jaspers, I.; Essers, J.; de Waard, H.; van Drunen, E.; Hanada, K.; Beverloo, B.; Hendriks, R.W.; de Klein, A.; Kanaar, R.; et al. Mutation of the mouse Rad17 gene leads to embryonic lethality and reveals a role in DNA damage-dependent recombination. EMBO J. 2004, 23, 3548–3558. [Google Scholar] [CrossRef]
- Pandita, R.K.; Sharma, G.G.; Laszlo, A.; Hopkins, K.M.; Davey, S.; Chakhparonian, M.; Gupta, A.; Wellinger, R.J.; Zhang, J.; Powell, S.N.; et al. Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol. Cell. Biol. 2006, 26, 1850–1864. [Google Scholar] [CrossRef]
- Bailey, C.; Fryer, A.E.; Greenslade, M. Warsaw Breakage Syndrome—A further report, emphasising cutaneous findings. Eur. J. Med. Genet. 2015, 58, 235–237. [Google Scholar] [CrossRef]
- Capo-Chichi, J.M.; Bharti, S.K.; Sommers, J.A.; Yammine, T.; Chouery, E.; Patry, L.; Rouleau, G.A.; Samuels, M.E.; Hamdan, F.F.; Michaud, J.L.; et al. Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw breakage syndrome. Hum. Mutat. 2013, 34, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Eppley, S.; Hopkin, R.J.; Mendelsohn, B.; Slavotinek, A.M. Clinical Report: Warsaw Breakage Syndrome with small radii and fibulae. Am. J. Med. Genet. A 2017, 173, 3075–3081. [Google Scholar] [CrossRef] [PubMed]
- Van der Lelij, P.; Chrzanowska, K.H.; Godthelp, B.C.; Rooimans, M.A.; Oostra, A.B.; Stumm, M.; Zdzienicka, M.Z.; Joenje, H.; de Winter, J.P. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 2010, 86, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Ooka, M.; Kawasumi, R.; Miyata, K.; Takata, M.; Hirota, K.; Branzei, D. Warsaw breakage syndrome DDX11 helicase acts jointly with RAD17 in the repair of bulky lesions and replication through abasic sites. Proc. Natl. Acad. Sci. USA 2018, 115, 8412–8417. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiedz, W.; Mosedale, G.; Johnson, M.; Ong, C.Y.; Pace, P.; Patel, K.J. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol. Cell 2004, 15, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Centore, R.C.; Yazinski, S.A.; Tse, A.; Zou, L. Spartan/C1orf124, a reader of PCNA ubiquitylation and a regulator of UV-induced DNA damage response. Mol. Cell 2012, 46, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, G.; Leung, J.W.; Nair, B.C.; Fong, K.W.; Chen, J. Proliferating cell nuclear antigen (PCNA)-binding protein C1orf124 is a regulator of translesion synthesis. J. Biol. Chem. 2012, 287, 34225–34233. [Google Scholar] [CrossRef]
- Juhasz, S.; Balogh, D.; Hajdu, I.; Burkovics, P.; Villamil, M.A.; Zhuang, Z.; Haracska, L. Characterization of human Spartan/C1orf124, an ubiquitin-PCNA interacting regulator of DNA damage tolerance. Nucleic Acids Res. 2012, 40, 10795–10808. [Google Scholar] [CrossRef] [Green Version]
- Machida, Y.; Kim, M.S.; Machida, Y.J. Spartan/C1orf124 is important to prevent UV-induced mutagenesis. Cell Cycle 2012, 11, 3395–3402. [Google Scholar] [CrossRef] [Green Version]
- Toth, A.; Hegedus, L.; Juhasz, S.; Haracska, L.; Burkovics, P. The DNA-binding box of human SPARTAN contributes to the targeting of Poleta to DNA damage sites. DNA Repair 2017, 49, 33–42. [Google Scholar] [CrossRef]
- Davis, E.J.; Lachaud, C.; Appleton, P.; Macartney, T.J.; Nathke, I.; Rouse, J. DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat. Struct. Mol. Biol. 2012, 19, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Mosbech, A.; Gibbs-Seymour, I.; Kagias, K.; Thorslund, T.; Beli, P.; Povlsen, L.; Nielsen, S.V.; Smedegaard, S.; Sedgwick, G.; Lukas, C.; et al. DVC1 (C1orf124) is a DNA damage-targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat. Struct. Mol. Biol. 2012, 19, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Maskey, R.S.; Flatten, K.S.; Sieben, C.J.; Peterson, K.L.; Baker, D.J.; Nam, H.J.; Kim, M.S.; Smyrk, T.C.; Kojima, Y.; Machida, Y.; et al. Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res. 2017, 45, 4564–4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morocz, M.; Zsigmond, E.; Toth, R.; Enyedi, M.Z.; Pinter, L.; Haracska, L. DNA-dependent protease activity of human Spartan facilitates replication of DNA-protein crosslink-containing DNA. Nucleic Acids Res. 2017, 45, 3172–3188. [Google Scholar] [CrossRef] [PubMed]
- Stingele, J.; Bellelli, R.; Alte, F.; Hewitt, G.; Sarek, G.; Maslen, S.L.; Tsutakawa, S.E.; Borg, A.; Kjaer, S.; Tainer, J.A.; et al. Mechanism and regulation of DNA-protein crosslink repair by the DNA-dependent metalloprotease SPRTN. Mol. Cell 2016, 64, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Vaz, B.; Popovic, M.; Newman, J.A.; Fielden, J.; Aitkenhead, H.; Halder, S.; Singh, A.N.; Vendrell, I.; Fischer, R.; Torrecilla, I.; et al. Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-protein crosslink repair. Mol. Cell 2016, 64, 704–719. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, A.; Kajita, K.; Ooka, M.; Akagawa, R.; Abe, T.; Takeda, S.; Branzei, D.; Hirota, K. SPARTAN promotes genetic diversification of the immunoglobulin-variable gene locus in avian DT40 cells. DNA Repair 2018, 68, 50–57. [Google Scholar] [CrossRef]
- Lee, K.Y.; Myung, K. PCNA modifications for regulation of post-replication repair pathways. Mol. Cells 2008, 26, 5–11. [Google Scholar]
- McIntyre, J.; Woodgate, R. Regulation of translesion DNA synthesis: Posttranslational modification of lysine residues in key proteins. DNA Repair 2015, 29, 166–179. [Google Scholar] [CrossRef] [Green Version]
- Kanao, R.; Masutani, C. Regulation of DNA damage tolerance in mammalian cells by post-translational modifications of PCNA. Mutat. Res. 2017, 803–805, 82–88. [Google Scholar] [CrossRef]
- Simpson, L.J.; Ross, A.L.; Szuts, D.; Alviani, C.A.; Oestergaard, V.H.; Patel, K.J.; Sale, J.E. RAD18-independent ubiquitination of proliferating-cell nuclear antigen in the avian cell line DT40. EMBO Rep. 2006, 7, 927–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, T.; Sonoda, E.; Yoshimura, M.; Kawano, Y.; Saya, H.; Kohzaki, M.; Takeda, S. Multiple roles of vertebrate REV genes in DNA repair and recombination. Mol. Cell. Biol. 2005, 25, 6103–6111. [Google Scholar] [CrossRef] [PubMed]
- Edmunds, C.E.; Simpson, L.J.; Sale, J.E. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell 2008, 30, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Seki, M.; Enomoto, T. Rad18/Rad5/Mms2-mediated polyubiquitination of PCNA is implicated in replication completion during replication stress. Genes Cells 2004, 9, 1031–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hishida, T.; Kubota, Y.; Carr, A.M.; Iwasaki, H. RAD6-RAD18-RAD5-pathway-dependent tolerance to chronic low-dose ultraviolet light. Nature 2009, 457, 612–615. [Google Scholar] [CrossRef]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Torres-Ramos, C.A.; Prakash, S.; Prakash, L. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 2002, 22, 2419–2426. [Google Scholar] [CrossRef]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation regulates Rad18-mediated template switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef]
- Pilzecker, B.; Buoninfante, O.A.; van den Berk, P.; Lancini, C.; Song, J.Y.; Citterio, E.; Jacobs, H. DNA damage tolerance in hematopoietic stem and progenitor cells in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E6875–E6883. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Poe, J.C.; Yang, L.; Fedoriw, A.; Desai, S.; Magnuson, T.; Li, Z.; Fedoriw, Y.; Araki, K.; Gao, Y.; et al. Rad18 confers hematopoietic progenitor cell DNA damage tolerance independently of the Fanconi Anemia pathway in vivo. Nucleic Acids Res. 2016, 44, 4174–4188. [Google Scholar] [CrossRef]
- Huang, J.; Huen, M.S.; Kim, H.; Leung, C.C.; Glover, J.N.; Yu, X.; Chen, J. RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat. Cell Biol. 2009, 11, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motegi, A.; Liaw, H.J.; Lee, K.Y.; Roest, H.P.; Maas, A.; Wu, X.; Moinova, H.; Markowitz, S.D.; Ding, H.; Hoeijmakers, J.H.; et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. USA 2008, 105, 12411–12416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motegi, A.; Sood, R.; Moinova, H.; Markowitz, S.D.; Liu, P.P.; Myung, K. Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. J. Cell Biol. 2006, 175, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Unk, I.; Hajdu, I.; Blastyak, A.; Haracska, L. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair 2010, 9, 257–267. [Google Scholar] [CrossRef]
- Unk, I.; Hajdu, I.; Fatyol, K.; Szakal, B.; Blastyak, A.; Bermudez, V.; Hurwitz, J.; Prakash, L.; Prakash, S.; Haracska, L. Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 18107–18112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomi, N.S.; Davari, K.; Grotzky, D.; Loos, F.; Bottcher, K.; Frankenberger, S.; Jungnickel, B. Analysis of SHPRH functions in DNA repair and immunoglobulin diversification. DNA Repair 2014, 24, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Durocher, D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair 2009, 8, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Scully, R.; Ganesan, S.; Vlasakova, K.; Chen, J.; Socolovsky, M.; Livingston, D.M. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol. Cell 1999, 4, 1093–1099. [Google Scholar] [CrossRef]
- Zhao, G.Y.; Sonoda, E.; Barber, L.J.; Oka, H.; Murakawa, Y.; Yamada, K.; Ikura, T.; Wang, X.; Kobayashi, M.; Yamamoto, K.; et al. A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol. Cell 2007, 25, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Keka, I.S.; Guilbaud, G.; Sale, J.; Narita, T.; Abdel-Aziz, H.I.; Wang, X.; Ogawa, S.; Sasanuma, H.; Chiu, R.; et al. The role of HERC2 and RNF8 ubiquitin E3 ligases in the promotion of translesion DNA synthesis in the chicken DT40 cell line. DNA Repair 2016, 40, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, H.; Hauschild, J.; Buerstedde, J.M. Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science 2002, 295, 1301–1306. [Google Scholar] [CrossRef]
- Oka, H.; Sakai, W.; Sonoda, E.; Nakamura, J.; Asagoshi, K.; Wilson, S.H.; Kobayashi, M.; Yamamoto, K.; Heierhorst, J.; Takeda, S.; et al. DNA damage response protein ASCIZ links base excision repair with immunoglobulin gene conversion. Biochem. Biophys. Res. Commun. 2008, 371, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Budzynska, P.M.; Kylaniemi, M.K.; Kallonen, T.; Soikkeli, A.I.; Nera, K.P.; Lassila, O.; Alinikula, J. Bach2 regulates AID-mediated immunoglobulin gene conversion and somatic hypermutation in DT40 B cells. Eur. J. Immunol. 2017, 47, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Abdel-Aziz, H.I.; Taniguchi, Y.; Yamazoe, M.; Takeda, S.; Hirota, K. Bloom DNA helicase facilitates homologous recombination between diverged homologous sequences. J. Biol. Chem. 2009, 284, 26360–26367. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G.; Ganesh, K.; Xue, K.; Lu, M.; Rada, C.; Neuberger, M.S. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol. Cell 2008, 31, 474–484. [Google Scholar] [CrossRef]
- Kitao, H.; Kimura, M.; Yamamoto, K.; Seo, H.; Namikoshi, K.; Agata, Y.; Ohta, K.; Takata, M. Regulation of histone H4 acetylation by transcription factor E2A in Ig gene conversion. Int. Immunol. 2008, 20, 277–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef]
- Kitao, H.; Nanda, I.; Sugino, R.P.; Kinomura, A.; Yamazoe, M.; Arakawa, H.; Schmid, M.; Innan, H.; Hiom, K.; Takata, M. FancJ/Brip1 helicase protects against genomic losses and gains in vertebrate cells. Genes Cells 2011, 16, 714–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, K.; Taniguchi, Y.; Hatanaka, A.; Sonoda, E.; Hochegger, H.; Adachi, N.; Matsuzaki, Y.; Koyama, H.; van Gent, D.C.; Jasin, M.; et al. Fen-1 facilitates homologous recombination by removing divergent sequences at DNA break ends. Mol. Cell. Biol. 2005, 25, 6948–6955. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Hashimoto, S.; Seo, H.; Shibata, T.; Ohta, K. Modulation of immunoglobulin gene conversion frequency and distribution by the histone deacetylase HDAC2 in chicken DT40. Genes Cells 2008, 13, 255–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, L.J.; Sale, J.E. UBE2V2 (MMS2) is not required for effective immunoglobulin gene conversion or DNA damage tolerance in DT40. DNA Repair 2005, 4, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Campo, V.A.; Patenaude, A.M.; Kaden, S.; Horb, L.; Firka, D.; Jiricny, J.; Di Noia, J.M. MSH6- or PMS2-deficiency causes re-replication in DT40 B cells, but it has little effect on immunoglobulin gene conversion or on repair of AID-generated uracils. Nucleic Acids Res. 2013, 41, 3032–3046. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, M.; Sonoda, E.; Nojima, K.; Sale, J.E.; Takenaka, K.; Kikuchi, K.; Taniguchi, Y.; Nakamura, K.; Sumitomo, Y.; Bree, R.T.; et al. Genetic evidence for single-strand lesions initiating Nbs1-dependent homologous recombination in diversification of Ig V in chicken B lymphocytes. PLoS Genet. 2009, 5, e1000356. [Google Scholar] [CrossRef] [PubMed]
- Paddock, M.N.; Buelow, B.D.; Takeda, S.; Scharenberg, A.M. The BRCT domain of PARP-1 is required for immunoglobulin gene conversion. PLoS Biol. 2010, 8, e1000428. [Google Scholar] [CrossRef] [PubMed]
- Hosono, Y.; Abe, T.; Ishiai, M.; Islam, M.N.; Arakawa, H.; Wang, W.; Takeda, S.; Ishii, Y.; Takata, M.; Seki, M.; et al. Tumor suppressor RecQL5 controls recombination induced by DNA crosslinking agents. Biochim. Biophys. Acta 2014, 1843, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, L.J.; Sale, J.E. Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. EMBO J. 2003, 22, 1654–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
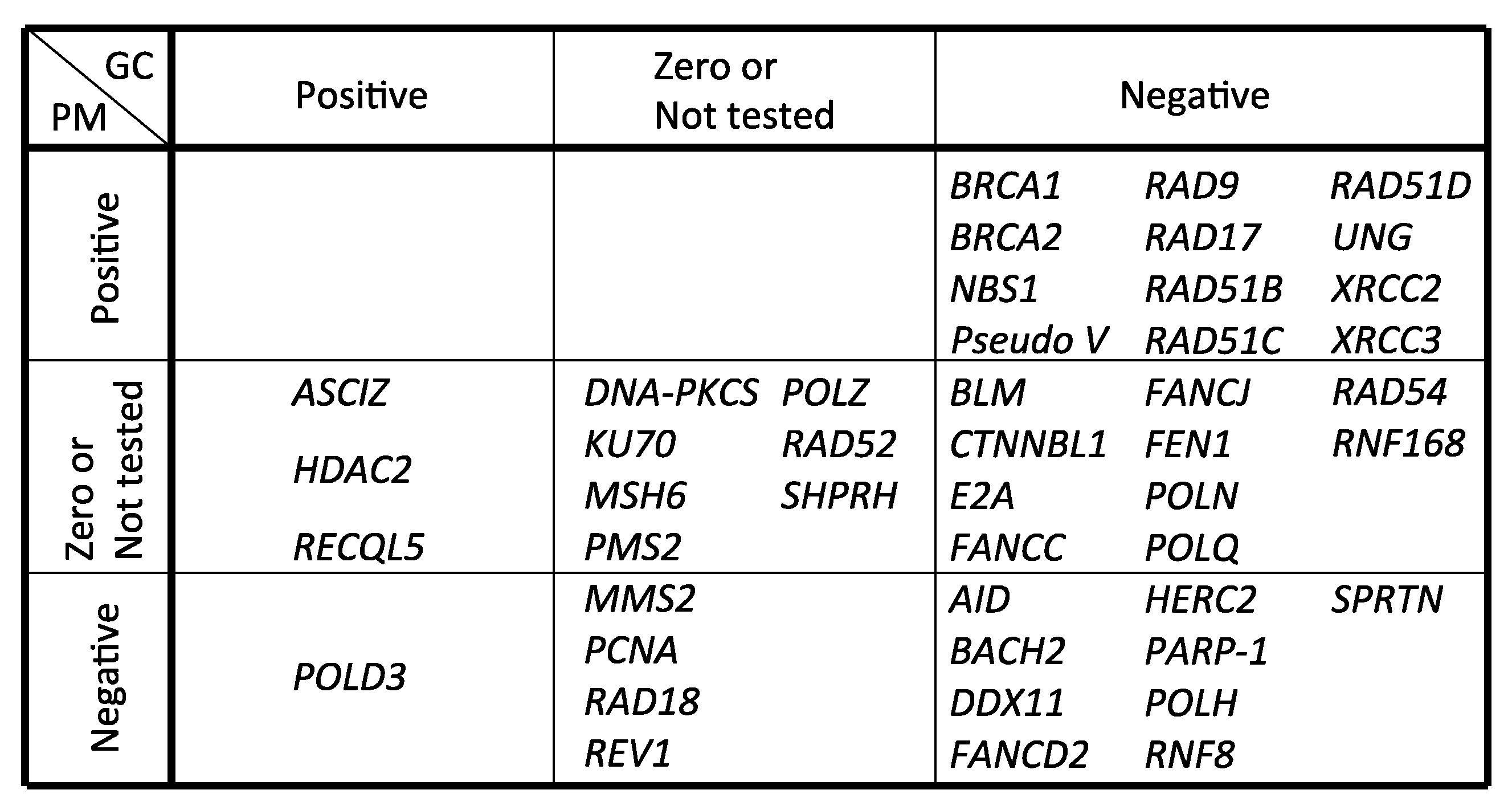
| Gene Name | GC | PM | Method | Reference |
|---|---|---|---|---|
| AID | − | − | IgM gain and sequencing | Arakawa et al., 2002 [105] |
| ASCIZ | + | 0 | IgM gain, mAID overexpression and sequencing | Oka et al., 2008 [106] |
| BACH2 | − | − | IgM gain and sequencing | Budzynska et al., 2017 [107] |
| BLM | − | NT | IgM gain | Kikuchi et al., 2009 [108] |
| BRCA1 | − | + | IgM loss and sequencing | Longerich et al., 2008 [42] |
| BRCA2 | − | + | IgM loss and sequencing | Hatanaka et al., 2005 [43] |
| CTNNBL1 | − | NT | IgM gain | Conticello et al., 2008 [109] |
| DDX11 | − | − | IgM gain, mAID overexpression and sequencing | Abe et al., 2018 [65] |
| DNA-PKCS | NT | 0 | IgM loss | Sale et al., 2001 [40] |
| E2A | − | 0 | IgM gain and sequencing | Kitao et al., 2008 [110] |
| FANCC | − | NT | IgM loss and sequencing | Pace et al., 2010 [111] |
| FANCD2 | − | − | IgM loss, gain and sequencing | Yamamoto et al., 2005 [45] |
| FANCJ | − | NT | IgM loss and sequencing | Kitao et al., 2011 [112] |
| FEN1 | − | NT | IgM gain and sequencing | Kikuchi et al., 2005 [113] |
| HDAC2 | + | 0 | IgM gain and sequencing | Lin et al., 2008 [114] |
| HERC2 | − | − | mAID overexpression and sequencing | Mohiuddin et al., 2016 [104] |
| KU70 | NT | 0 | IgM loss | Sale et al., 2001 [40] |
| MMS2 | 0 | − | IgM loss and sequencing | Simpson et al., 2005 [115] |
| MSH6 | 0 | 0 | IgM gain and sequencing | Campo et al., 2013 [116] |
| NBS1 (p70) | − | + | sIgM gain, mAID overexpression and sequencing | Nakahara et al., 2009 [117] |
| PARP-1 | − | − | IgM gain, mAID overexpression and sequencing | Paddock et al., 2010 [118] |
| PCNA (K164R) | NT | − | IgM loss and sequencing | Arakawa et al., 2006 [27] |
| PMS2 | 0 | 0 | IgM gain and sequencing | Campo et al., 2013 [116] |
| POLD3 | + | − | mAID overexpression and sequencing | Hirota et al., 2015 [16] |
| POLH | − | − | IgM gain and sequencing | Kawamoto et al., 2006 [46], Kohzaki et al., 2010 [17] |
| POLN | − | 0 | IgM gain, mAID overexpression and sequencing | Kohzaki et al., 2010 [17] |
| POLQ | − | 0 | IgM gain, mAID overexpression and sequencing | Kohzaki et al., 2010 [17] |
| POLH/POLN/POLQ | − | − | IgM gain, mAID overexpression and sequencing | Kohzaki et al., 2010 [17] |
| POLZ (REV3) | 0 | NT | IgM gain and sequencing | Okada et al., 2005 [83] |
| Pseudo V | − | + | IgM loss and sequencing | Arakawa et al., 2004 [28] |
| RAD9 | − | + | mAID overexpression and sequencing | Saberi et al., 2008 [38] |
| RAD17 | − | + | mAID overexpression and sequencing | Saberi et al., 2008 [38] |
| RAD18 | NT | − | IgM loss and sequencing | Arakawa et al., 2006 [27] |
| RAD51B | − | + | IgM loss and sequencing | Sale et al., 2001 [40] |
| RAD51C | − | + | IgM loss and sequencing | Hatanaka et al., 2005 [43] |
| RAD51D | − | + | IgM loss and sequencing | Hatanaka et al., 2005 [43] |
| RAD52 | NT | 0 | IgM loss | Sale et al., 2001 [40] |
| RAD54 | − | NT | IgM gain and sequencing | Bezzubova et al., 1997 [44] |
| RECQL5 | + | NT | IgM gain and sequencing | Hosono et al., 2014 [119] |
| RNF8 | − | − | mAID overexpression and sequencing | Mohiuddin et al., 2016 [104] |
| RNF168 | − | 0 | mAID overexpression and sequencing | Mohiuddin et al., 2016 [104] |
| SPRTN | − | − | IgM gain, mAID overexpression and sequencing | Nakazato et al., 2018 [78] |
| SHPRH | 0 | 0 | sIgM gain and sequencing | Tomi et al., 2014 [97] |
| REV1 | 0 | − | IgM loss and sequencing | Simpson et al., 2003 [120] |
| UNG | − | + | IgM gain, loss and sequencing | Saribasak et al., 2005 [25] |
| XRCC2 | − | + | IgM loss and sequencing | Sale et al., 2001 [40] |
| XRCC3 | − | + | IgM loss and sequencing | Sale et al., 2001 [40] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abe, T.; Branzei, D.; Hirota, K. DNA Damage Tolerance Mechanisms Revealed from the Analysis of Immunoglobulin V Gene Diversification in Avian DT40 Cells. Genes 2018, 9, 614. https://doi.org/10.3390/genes9120614
Abe T, Branzei D, Hirota K. DNA Damage Tolerance Mechanisms Revealed from the Analysis of Immunoglobulin V Gene Diversification in Avian DT40 Cells. Genes. 2018; 9(12):614. https://doi.org/10.3390/genes9120614
Chicago/Turabian StyleAbe, Takuya, Dana Branzei, and Kouji Hirota. 2018. "DNA Damage Tolerance Mechanisms Revealed from the Analysis of Immunoglobulin V Gene Diversification in Avian DT40 Cells" Genes 9, no. 12: 614. https://doi.org/10.3390/genes9120614
APA StyleAbe, T., Branzei, D., & Hirota, K. (2018). DNA Damage Tolerance Mechanisms Revealed from the Analysis of Immunoglobulin V Gene Diversification in Avian DT40 Cells. Genes, 9(12), 614. https://doi.org/10.3390/genes9120614