Microbiome Data Accurately Predicts the Postmortem Interval Using Random Forest Regression Models

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Amplicon Sequencing Data Processing
2.2. Assigning Taxonomy
2.3. Model Testing
3. Results
3.1. Cross-validation Error Rates
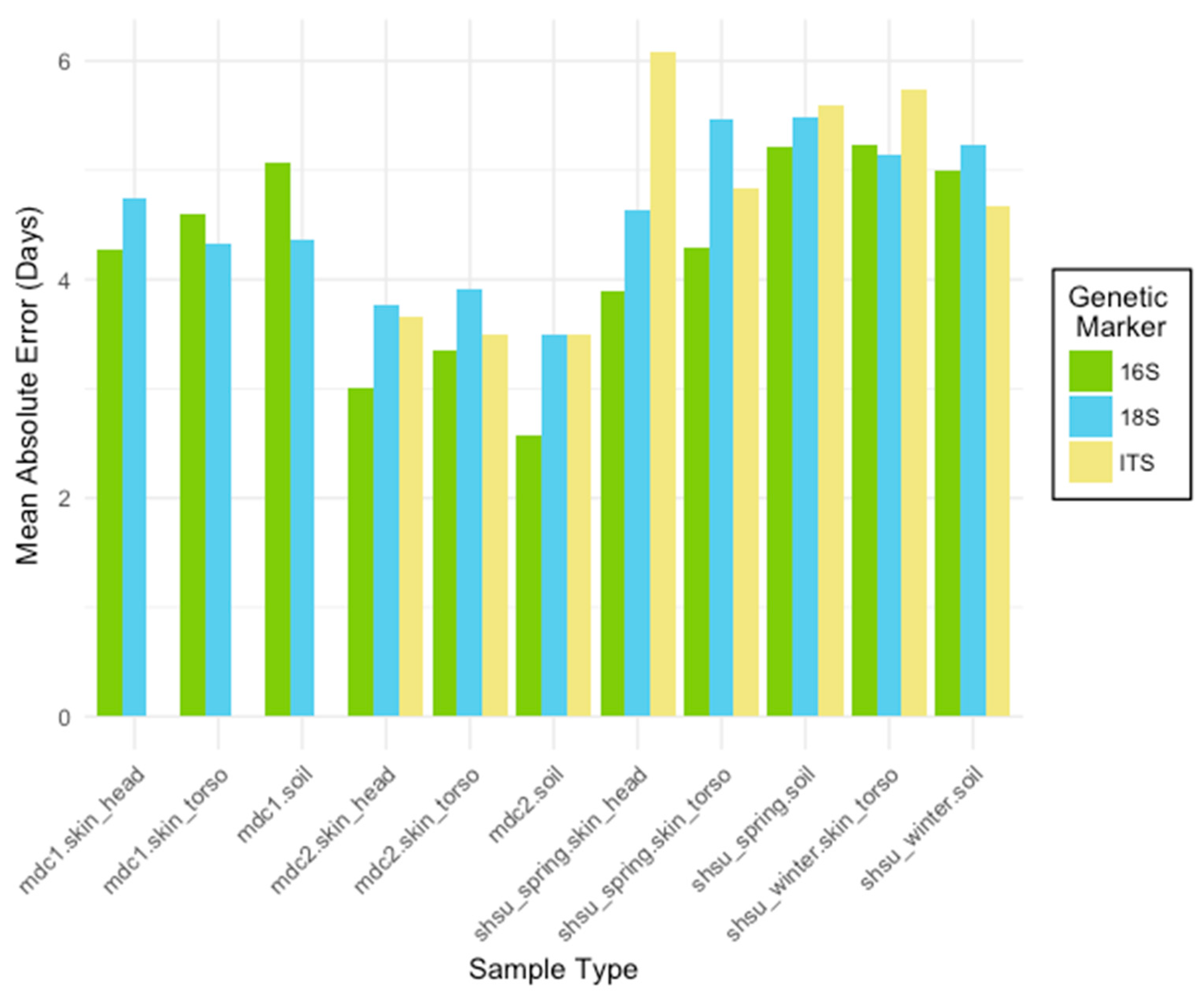
3.1.1. Comparison of Sample Types
3.1.2. Comparison of Genetic Markers across Sequence Variant and Taxonomic Levels
3.2. Cross-study Error Rates
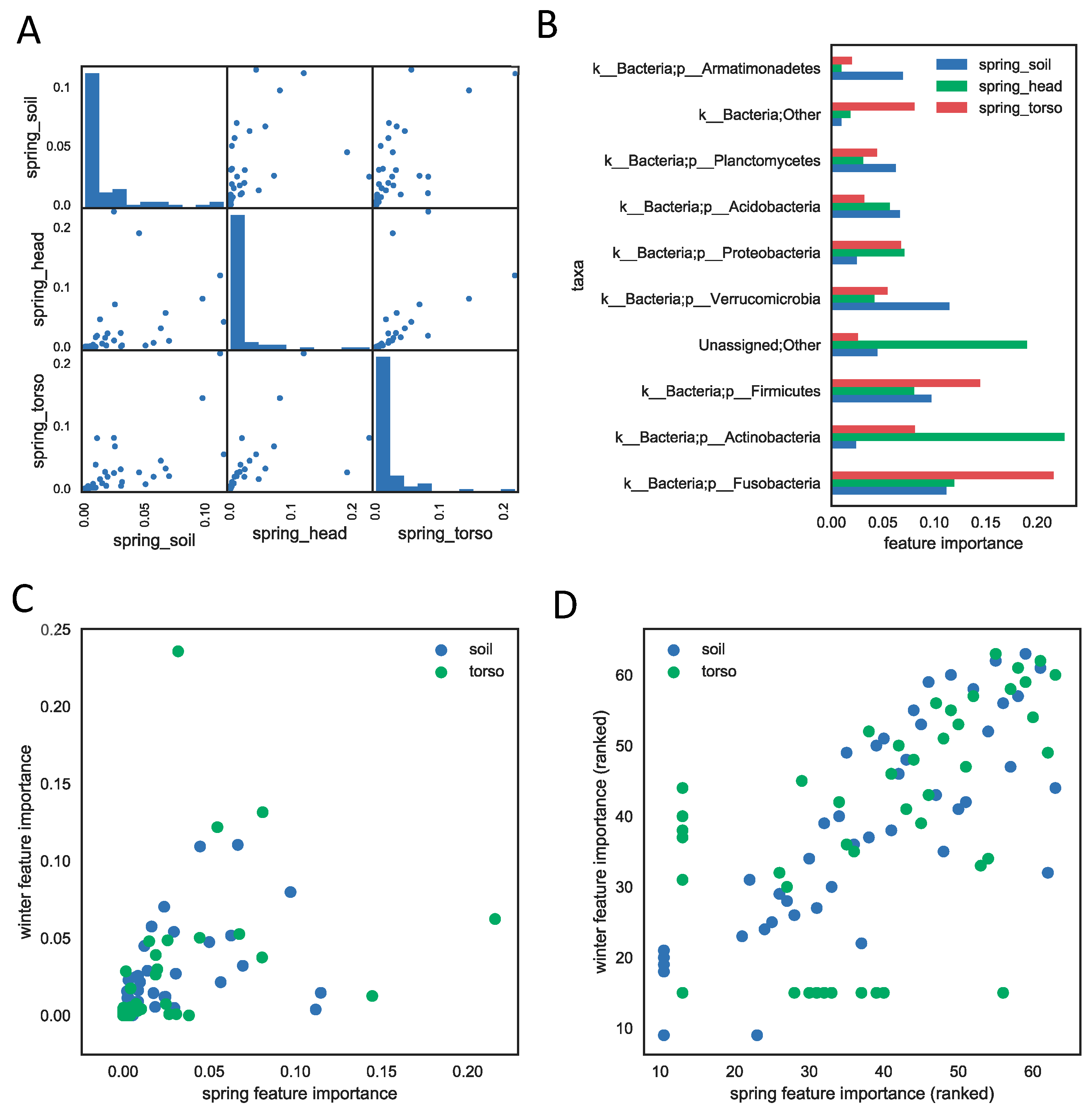
3.3. Important Feature Taxa
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McGeachie, M.J.; Sordillo, J.E.; Gibson, T.; Weinstock, G.M.; Liu, Y.-Y.; Gold, D.R.; Weiss, S.T.; Litonjua, A. Longitudinal prediction of the infant gut microbiome with dynamic bayesian networks. Sci. Rep. 2016, 6, 20359. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.O.; Yellowlees, D.; Tibbett, M. Cadaver decomposition in terrestrial ecosystems. Die Naturwiss. 2007, 94, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Noronha, M.F.; Lacerda Júnior, G.V.; Gilbert, J.A.; de Oliveira, V.M. Taxonomic and functional patterns across soil microbial communities of global biomes. Sci. Total Environ. 2017, 609, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Jansson, J.K.; Gilbert, J.A.; Knight, R.; Consortium, T.E.M.P. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Parfrey, L.W.; Walters, W.A.; Lauber, C.L.; Clemente, J.C.; Berg-Lyons, D.; Teiling, C.; Kodira, C.; Mohiuddin, M.; Brunelle, J.; Driscoll, M.; et al. Communities of microbial eukaryotes in the mammalian gut within the context of environmental eukaryotic diversity. Front. Microbiol. 2014, 5, 298. [Google Scholar] [CrossRef] [PubMed]
- Willger, S.D.; Grim, S.L.; Dolben, E.L.; Shipunova, A.; Hampton, T.H.; Morrison, H.G.; Filkins, L.M.; O’Toole, G.A.; Moulton, L.A.; Ashare, A.; et al. Characterization and quantification of the fungal microbiome in serial samples from individuals with cystic fibrosis. Microbiome 2014, 2, 40. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, K.S.; Leff, J.W.; Barberán, A.; Bates, S.T.; Betley, J.; Crowther, T.W.; Kelly, E.F.; Oldfield, E.E.; Shaw, E.A.; Steenbock, C.; et al. Biogeographic patterns in below-ground diversity in New York City’s Central Park are similar to those observed globally. Proc. R. Soc. B Biol. Sci. 2014, 281. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, J.L.; Xu, Z.Z.; Bouslimani, A.; Dorrestein, P.; Carter, D.O.; Knight, R. Microbiome tools for forensic science. Trends Biotechnol. 2017, 1482, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Amendt, J.; Campobasso, C.P.; Gaudry, E.; Reiter, C.; LeBlanc, H.N.; Hall, M.J. Best practice in forensic entomology—Standards and guidelines. Int. J. Leg. Med. 2007, 121, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, J.L.; Wegener Parfrey, L.; Gonzalez, A.; Lauber, C.L.; Knights, D.; Ackermann, G.; Humphrey, G.C.; Gebert, M.J.; Van Treuren, W.; Berg-Lyons, D.; et al. A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system. eLife 2013, 2, e01104. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, J.L.; Xu, Z.Z.; Weiss, S.; Lax, S.; Van Treuren, W.; Hyde, E.R.; Song, S.J.; Amir, A.; Larsen, P.; Sangwan, N.; et al. Microbial community assembly and metabolic function during mammalian corpse decomposition. Science 2016, 351, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Pechal, J.L.; Crippen, T.L.; Tarone, A.M.; Lewis, A.J.; Tomberlin, J.K.; Benbow, M.E. Microbial community functional change during vertebrate carrion decomposition. PLoS ONE 2013, 8, e79035. [Google Scholar] [CrossRef] [PubMed]
- Hauther, K.A.; Cobaugh, K.L.; Jantz, L.M.; Sparer, T.E.; DeBruyn, J.M. Estimating time since death from postmortem human gut microbial communities. J. Forensic Sci. 2015, 60, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- DeBruyn, J.M.; Hauther, K.A. Postmortem succession of gut microbial communities in deceased human subjects. PeerJ 2017, 5, e3437. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.R.; Trinidad, D.D.; Guzman, S.; Khan, Z.; Parziale, J.V.; DeBruyn, J.M.; Lents, N.H. A machine learning approach for using the postmortem skin microbiome to estimate the postmortem interval. PLoS ONE 2016, 11, e0167370. [Google Scholar] [CrossRef] [PubMed]
- Cobaugh, K.L.; Schaeffer, S.M.; DeBruyn, J.M. Functional and structural succession of soil microbial communities below decomposing human cadavers. PLoS ONE 2015, 10, e0130201. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.O.; Metcalf, J.L.; Bibat, A.; Knight, R. Seasonal variation of postmortem microbial communities. Forensic Sci. Med. Pathol. 2015, 11, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Carter, D.O.; Metcalf, J.L.; Knight, R. Carcass mass has little influence on the structure of gravesoil microbial communities. Int. J. Leg. Med. 2016, 130, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Costello, E.K.; Knight, R. Supervised classification of human microbiota. FEMS Microbiol. Rev. 2011, 35, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L.; Friedman, J.H.; Olshen, R.A.; Stone, C.J. Classification and Regression Trees, 1993; Chapman Hall: New York, NY, USA, 1984. [Google Scholar]
- Zhang, G.; Lu, Y. Bias-corrected random forests in regression. J. Appl. Stat. 2012, 39, 151–160. [Google Scholar] [CrossRef]
- QIITA. Available online: https://qiita.ucsd.edu.
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur rapidly resolves single-nucleotide community sequence patterns. MSystems 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.; Xu, Z.Z.; Sanders, J.; Zhu, Q.; Bletz, M.C.; Tripathi, A.; Knight, R. Microbiome analysis with Calour. In publication.
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Louppe, G.; Prettenhofer, P.; Weiss, R.; et al. Scikit-learn: Machine Learning in Python. JMLR 2011, 12, 2825–2830. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing, 3.4.1; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Droettboom, M.; Caswell, T.A.; Hunter, J.; Firing, E.; Hedegaard Nielsen, J.; Root, B.; Elson, P.; Dale, D.; Lee, J.-J.; Varoquaux, N.; et al. Matplotlib/matplotlib: V2.0.0 (Version v2.0.0). Zenodo 2017. [Google Scholar] [CrossRef]
- Yazdani, M.; Taylor, B.C.; Debelius, J.W.; Li, W.; Knight, R.; Smarr, L. Using machine learning to identify major shifts in human gut microbiome protein family abundance in disease. In Proceedings of the 2016 IEEE International Conference on Big Data (Big Data), Washington, DC, USA, 5–8 December 2016; pp. 1272–1280. [Google Scholar]
- Kuhn, M.; Johnson, K. Applied Predictive Modeling; Springer Science and Business: New York, NY, USA, 2013; ISBN 978-1461468486. [Google Scholar]
- Byard, R.W. Timing: The Achilles heel of forensic pathology. Forensic Sci. Med. Pathol. 2017, 13, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Madea, B. Methods for determining time of death. Forensic Sci. Med. Pathol. 2016, 12, 451–485. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.C.; Pozhitkov, A.E.; Noble, P.A. Accurate predictions of postmortem interval using linear regression analyses of gene meter expression data. Forensic Sci. Int. 2017, 275, 90–101. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| QIITA Study Number | QIITA Study Name | Our Study Name | Shorthand Name | Prep Number | Marker | Trim Length | OTU Table Type | Number of Days Sampled |
|---|---|---|---|---|---|---|---|---|
| 714 | A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system | Mouse Decomposition 1 | mdc1 | 769 | 16S | 90 bp | reference-hit.biom | 48 |
| 1889 | A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system—18S | Mouse Decomposition 1 | mdc1 | 1204 | 18S | 90 bp | all.biom | 48 |
| 10141 | Metcalf microbial community assembly and metabolic function during mammalian corpse decomposition | Mouse Decomposition 2 | mdc2 | 1265 | 16S | 90 bp | reference-hit.biom | 70 |
| 1038 | 18S | 90 bp | all.biom | 70 | ||||
| 345 | ITS | 100 bp | all.biom | 70 | ||||
| 10142 | Metcalf microbial community assembly and metabolic function during mammalian corpse decomposition Sam Houston State University (SHSU) winter | SHSU Winter | shsu_winter | 333 | 16S | 90 bp | reference-hit.biom | 132 |
| 1166 | 18S | 90 bp | all.biom | 132 | ||||
| 335 | ITS | 100 bp | all.biom | 132 | ||||
| 10143 | Metcalf microbial community assembly and metabolic function during mammalian corpse decomposition Sam Houston State University (SHSU) April 2012 exp. | SHSU Spring | shsu_spring | 1107 | 16S | 90 bp | reference-hit.biom | 82 |
| 1109 | 18S | 90 bp | all.biom | 82 | ||||
| 1110 | ITS | 100 bp | all.biom | 82 |
| Genomic Marker | Study Name | Sample Type | Sequence Variants | Species Level | Genus Level | Family Level | Order Level | Class Level | Phylum Level |
|---|---|---|---|---|---|---|---|---|---|
| 16S | mdc1 | soil | 5.068 | 4.528 | 4.439 | 4.574 | 4.596 | 4.308 | 4.565 |
| skin_torso | 4.602 | 3.744 | 3.577 | 3.353 | 3.889 | 4.377 | 4.070 | ||
| skin_head | 4.272 | 3.816 | 3.816 | 3.747 | 3.442 | 3.315 | 4.672 | ||
| mdc2 | soil | 2.571 | 1.943 | 1.955 | 1.911 | 2.062 | 1.971 | 1.737 | |
| skin_torso | 3.357 | 2.926 | 2.898 | 2.783 | 2.826 | 2.942 | 2.856 | ||
| skin_head | 3.001 | 2.383 | 2.379 | 2.340 | 2.467 | 2.369 | 2.405 | ||
| shsu_spring | soil | 5.225 | 3.594 | 3.632 | 3.660 | 3.966 | 3.868 | 3.877 | |
| skin_torso | 4.303 | 3.830 | 3.807 | 4.106 | 4.343 | 4.311 | 4.022 | ||
| skin_head | 3.890 | 3.506 | 3.385 | 3.577 | 3.342 | 2.940 | 3.006 | ||
| shsu_winter | soil | 4.985 | 3.922 | 3.980 | 3.947 | 3.848 | 4.026 | 3.783 | |
| skin_torso | 5.237 | 4.543 | 4.483 | 4.385 | 3.970 | 3.704 | 3.265 | ||
| 18S | mdc1 | soil | 4.370 | 3.125 | 3.072 | 3.135 | 2.813 | 2.942 | 2.733 |
| skin_torso | 4.333 | 3.821 | 3.447 | 3.030 | 3.549 | 2.702 | 4.521 | ||
| skin_head | 4.744 | 4.583 | 4.138 | 4.616 | 4.251 | 3.775 | 4.657 | ||
| mdc2 | soil | 3.505 | 3.237 | 3.208 | 3.107 | 3.221 | 3.043 | 3.330 | |
| skin_torso | 3.907 | 3.870 | 3.856 | 3.676 | 3.910 | 3.867 | 3.704 | ||
| skin_head | 3.772 | 3.761 | 3.575 | 3.725 | 3.665 | 3.819 | 3.912 | ||
| shsu_spring | soil | 5.486 | 4.654 | 4.459 | 4.283 | 3.837 | 3.400 | 3.264 | |
| skin_torso | 5.457 | 4.654 | 5.196 | 5.404 | 5.264 | 5.754 | 5.974 | ||
| skin_head | 4.645 | 4.571 | 4.370 | 5.148 | 5.028 | 4.763 | 5.218 | ||
| shsu_winter | soil | 5.239 | 4.429 | 4.442 | 4.239 | 4.042 | 3.449 | 3.504 | |
| skin_torso | 5.141 | 4.880 | 4.721 | 4.962 | 5.028 | 4.660 | 4.604 | ||
| ITS | mdc2 | soil | 3.497 | 3.169 | 3.157 | 2.957 | 2.941 | 2.820 | 2.797 |
| skin_torso | 3.505 | 3.237 | 3.211 | 3.083 | 3.023 | 2.597 | 3.036 | ||
| skin_head | 3.648 | 3.561 | 3.523 | 3.483 | 3.509 | 3.413 | 3.305 | ||
| shsu_spring | soil | 5.586 | 4.735 | 4.836 | 4.629 | 4.980 | 4.461 | 4.713 | |
| skin_torso | 4.837 | 4.671 | 4.563 | 4.688 | 4.786 | 4.860 | 5.500 | ||
| skin_head | 6.080 | 5.996 | 6.083 | 5.803 | 6.090 | 5.965 | 5.416 | ||
| shsu_winter | soil | 4.675 | 4.114 | 3.965 | 3.954 | 3.933 | 3.671 | 4.077 | |
| skin_torso | 5.726 | 5.702 | 5.662 | 5.608 | 5.565 | 5.575 | 5.610 |
| Genomic Marker | Training Dataset | Sample Type | Sequence Variants MAE | Species Level MAE | Genus Level MAE | Family Level MAE | Order Level MAE | Class Level MAE | Phylum Level MAE |
|---|---|---|---|---|---|---|---|---|---|
| 16S | Spring | soil | 88.693 | 57.929 | 59.251 | 57.045 | 56.936 | 55.367 | 48.686 |
| skin | 92.598 | 90.197 | 90.584 | 104.770 | 109.412 | 117.672 | 135.749 | ||
| Winter | soil | 109.482 | 91.406 | 91.295 | 91.857 | 91.025 | 88.849 | 83.312 | |
| skin | 120.764 | 129.695 | 130.763 | 122.418 | 124.701 | 123.043 | 108.737 | ||
| 18S | Spring | soil | 81.013 | 62.572 | 62.850 | 55.648 | 51.316 | 51.481 | 50.082 |
| skin | 88.155 | 93.173 | 85.754 | 89.676 | 91.793 | 72.846 | 67.242 | ||
| Winter | soil | 96.145 | 82.780 | 75.628 | 72.228 | 67.725 | 71.757 | 63.465 | |
| skin | 111.004 | 110.772 | 101.268 | 101.222 | 101.524 | 107.409 | 105.248 | ||
| ITS | Spring | soil | 111.806 | 94.797 | 94.742 | 93.504 | 85.282 | 80.856 | 58.359 |
| skin | 101.852 | 96.815 | 99.272 | 101.086 | 104.162 | 106.043 | 94.468 | ||
| Winter | soil | 104.775 | 99.360 | 96.392 | 96.604 | 93.564 | 87.709 | 81.623 | |
| skin | 114.027 | 110.865 | 107.026 | 113.294 | 117.302 | 115.937 | 87.274 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belk, A.; Xu, Z.Z.; Carter, D.O.; Lynne, A.; Bucheli, S.; Knight, R.; Metcalf, J.L. Microbiome Data Accurately Predicts the Postmortem Interval Using Random Forest Regression Models. Genes 2018, 9, 104. https://doi.org/10.3390/genes9020104
Belk A, Xu ZZ, Carter DO, Lynne A, Bucheli S, Knight R, Metcalf JL. Microbiome Data Accurately Predicts the Postmortem Interval Using Random Forest Regression Models. Genes. 2018; 9(2):104. https://doi.org/10.3390/genes9020104
Chicago/Turabian StyleBelk, Aeriel, Zhenjiang Zech Xu, David O. Carter, Aaron Lynne, Sibyl Bucheli, Rob Knight, and Jessica L. Metcalf. 2018. "Microbiome Data Accurately Predicts the Postmortem Interval Using Random Forest Regression Models" Genes 9, no. 2: 104. https://doi.org/10.3390/genes9020104
APA StyleBelk, A., Xu, Z. Z., Carter, D. O., Lynne, A., Bucheli, S., Knight, R., & Metcalf, J. L. (2018). Microbiome Data Accurately Predicts the Postmortem Interval Using Random Forest Regression Models. Genes, 9(2), 104. https://doi.org/10.3390/genes9020104