Deep-Coverage MPS Analysis of Heteroplasmic Variants within the mtGenome Allows for Frequent Differentiation of Maternal Relatives



Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and DNA Extraction
2.2. Long-Range Polymerase Chain Reaction
2.3. MiSeq MPS
2.4. Data Analysis
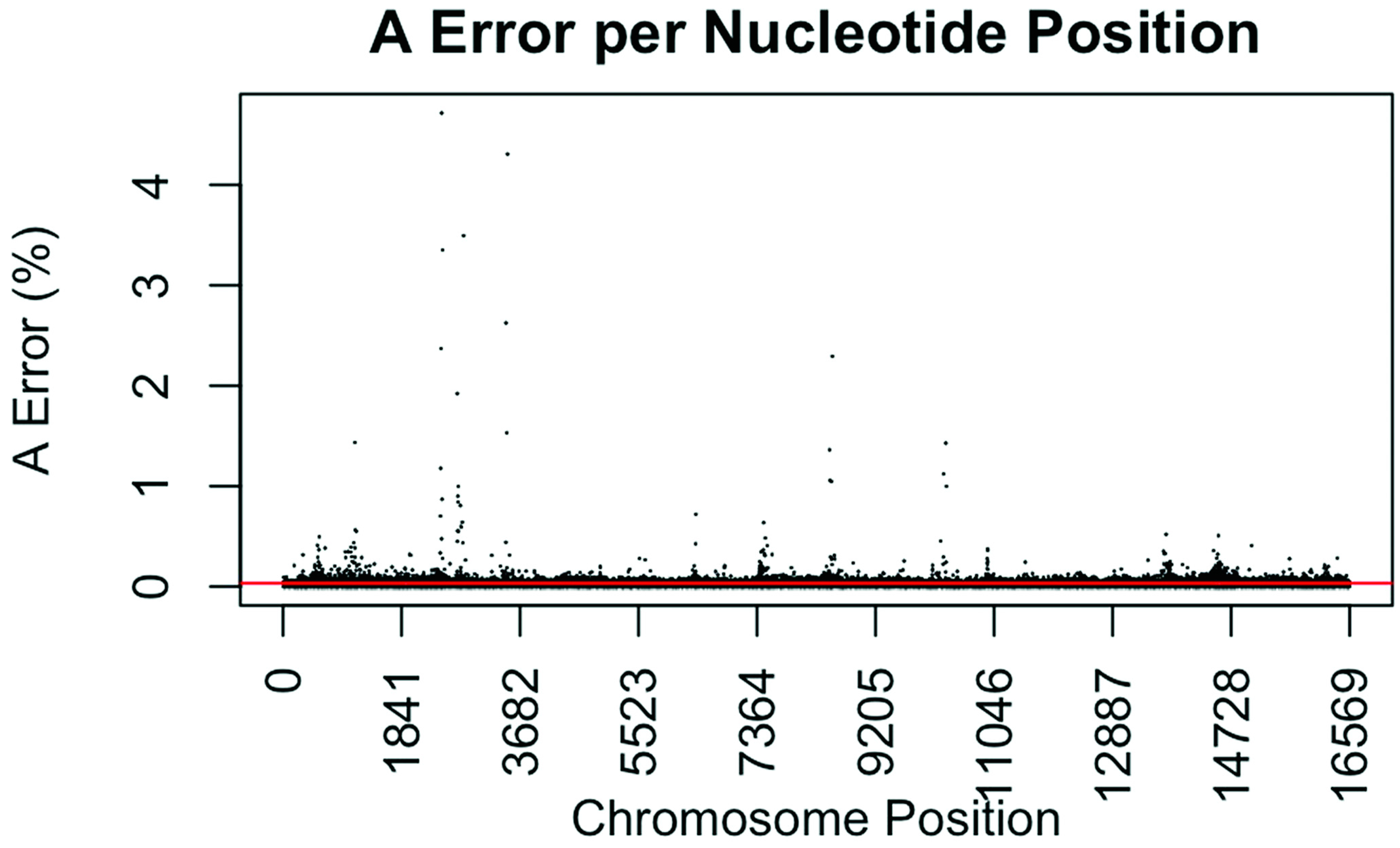
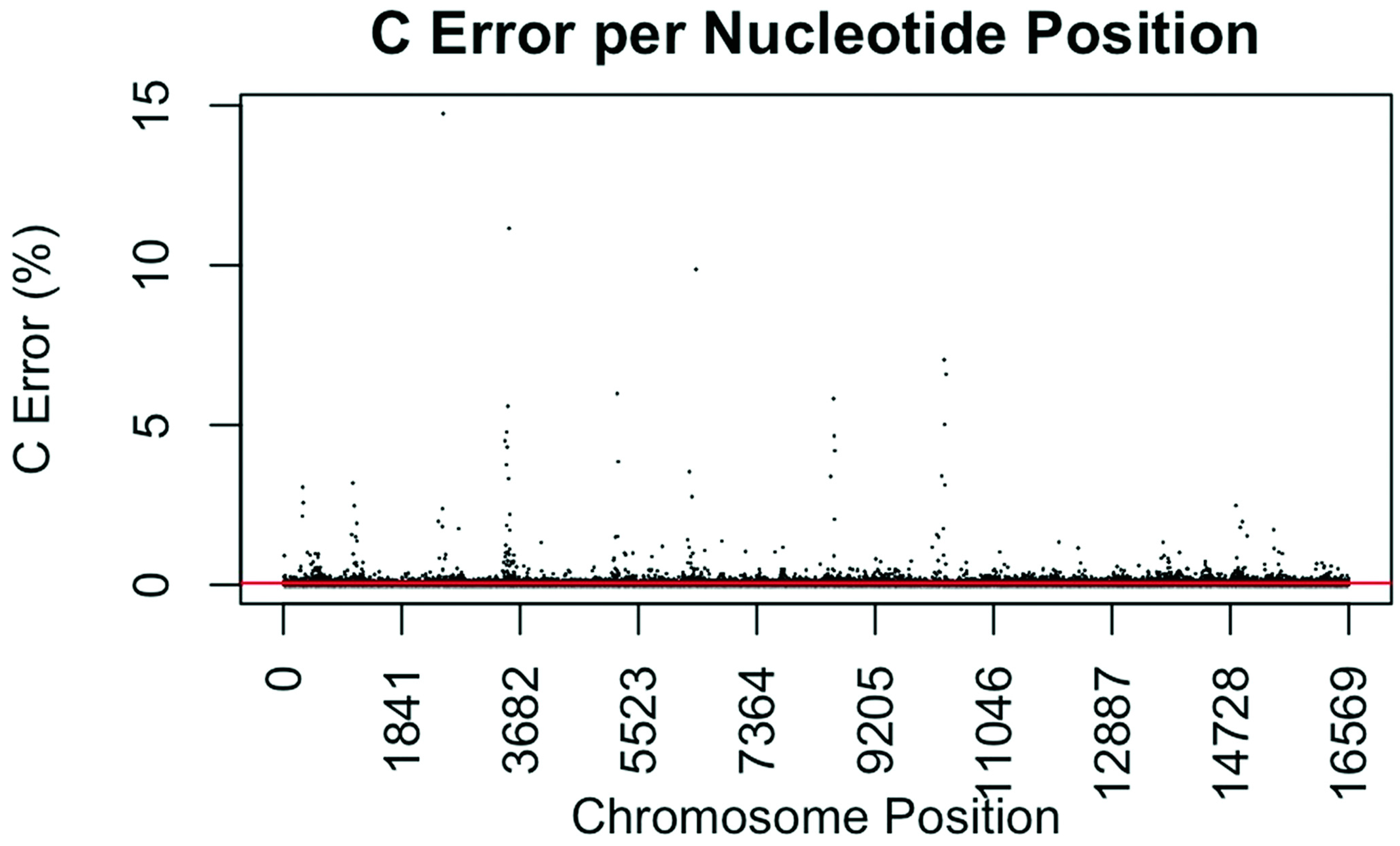
2.5. Error and Coverage Assessment
2.6. Haplogroup Generation
2.7. Statistical Analysis
3. Results and Discussion
3.1. Shared Heteroplasmy
3.2. Differentiating Heteroplasmy
3.3. Random Heteroplasmy
3.4. Coverage and Error Rates
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Bjenning, C.; Wang, F.; Wang, N.; Kream, R.M. Mitochondrial Heteroplasmy. Mitochondrial Dynamics in Cardiovascular Medicine, Advances in Experimental Medicine and Biology; Santulli, G., Ed.; Springer International Publishing: Cham, Switzerland, 2017; Volume 982, pp. 577–594. [Google Scholar] [CrossRef]
- Duggan, A.T.; Stoneking, M. A highly unstable recent mutation in human mtDNA. Am. J. Hum. Genet. 2013, 92, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Lopopolo, M.; Børsting, C.; Pereira, V.; Morling, N. A study of the peopling of Greenland using next generation sequencing of complete mitochondrial genomes. Am. J. Phys. Anthropol. 2016, 161, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Bodner, M.; Iuvaro, A.; Strobl, C.; Nagl, S.; Huber, G.; Pelotti, S.; Pettener, D.; Luiselli, D.; Parson, W. Helena, the hidden beauty: Resolving the most common West Eurasian mtDNA control region haplotype by massively parallel sequencing an Italian population sample. Forensic Sci. Int.-Genet. 2015, 15, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Gallimore, J.M.; McElhoe, J.A.; Holland, M.M. Assessing heteroplasmic variant drift in the mtDNA control region of human hairs using an MPS approach. Forensic Sci. Int.-Genet. 2018, 32, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial function, biology, and role in disease: A scientific statement from the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial bioenergetics etiology of disease. J. Clin. Invest. 2013, 123, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Balbi, V.; Balducci, E.; Pirazzini, C.; Rosini, F.; Tavano, F.; Achilli, A.; Siviero, P.; Minicuci, N.; Bellavista, E.; et al. Evidence for sub-haplogroup H5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Lièvre, A.; Chapusot, C.; Bouvier, A.M.; Zinzindohoué, F.; Piard, F.; Roignot, P.; Arnould, L.; Beaune, P.; Faivre, J.; Laurent-Puig, P. Clinical value of mitochondrial mutations in colorectal cancer. J. Clin. Oncol. 2005, 23, 3517–3525. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Invest. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 3, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo-Jaramillo, B.; Su, M.S.; Stoler, N.; McElhoe, J.A.; Dickins, B.; Blankenberg, D.; Korneliussen, T.S.; Chiaromonte, F.; Nielsen, R.; Holland, M.M.; et al. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 15474–15479. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Rothwell, R.; Vermaat, M.; Wachsmuth, M.; Schröder, R.; Laros, J.F.J.; van Oven, M.; de Bakker, P.I.W.; Bovenberg, J.A.; van Duijn, C.M.; et al. Transmission of human mtDNA heteroplasmy in the genome of the Netherlands families: Support for a variable-size bottleneck. Genome Res. 2016, 26, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.J.; Carling, P.J.; Alston, C.L.; Floros, V.I.; Pyle, A.; Hudson, G.; Sallevelt, S.C.E.H.; Lamperti, C.; Carelli, V.; Bindoff, L.A.; et al. Mitochondrial DNA sequence characteristics modulate the size of the genetic bottleneck. Hum. Mol. Genet. 2016, 25, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Ashley, M.V.; Laipis, P.J.; Hauswirth, W.W. Rapid segregation of heteroplasmic bovine mitochondria. Nuc. Acids Res. 1989, 17, 7325–7331. [Google Scholar] [CrossRef]
- Ivanov, P.L.; Wadhams, M.J.; Roby, R.K.; Holland, M.M.; Weedn, V.W.; Parsons, T.J. Mitochondrial DNA sequence heteroplasmy in the Grand Duke of Russia Georgji Romanov establishes the authenticity of the remains of Tsar Nicholas II. Nat. Genet. 1996, 12, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Cree, L.M.; Samuels, D.C.; de Sousa Lopes, S.C.; Rajasimha, H.K.; Wonnapinij, P.; Mann, J.R.; Dahl, H.H.; Chinnery, P.F. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 2008, 40, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Wonnapinij, P.; Chinnery, P.F.; Samuels, D.C. The distribution of mitochondrial DNA heteroplasmy due to random genetic drift. Am. J. Hum. Genet. 2008, 83, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Ivanov, P.L.; Kimpton, C.; Piercy, R.; Benson, N.; Tully, G.; Evett, I.; Hagelberg, E.; Sullivan, K. Identification of the remains of the Romanov family by DNA analysis. Nat. Genet. 1994, 6, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.A.; Saunier, J.L.; Niederstätter, H.; Strouss, K.M.; Sturk, K.A.; Diegoli, T.M.; Brandstätter, A.; Parson, W.; Parsons, T.J. Investigation of heteroplasmy in the human mitochondrial DNA control region: A synthesis of observations from more than 5000 global population samples. J. Mol. Evol. 2009, 68, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Parsons, T.J.; Muniec, D.S.; Sullivan, K.; Woodyatt, N.; Alliston-Greiner, R.; Wilson, M.R.; Berry, D.L.; Holland, K.A.; Weedn, V.W.; Gill, P.; et al. A high observed substitution rate in the human mitochondrial DNA control region. Nat Genet. 1997, 15, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Dickins, B.; Afgan, E.; Paul, I.M.; Taylor, J.; Makova, K.D.; Nekrutenko, A. Dynamics of mitochondrial heteroplasmy in three families investigated via a repeatable re-sequencing study. Genome Biol. 2011, 12, R59. [Google Scholar] [CrossRef] [PubMed]
- Pallotti, F.; Binelli, G.; Fabbri, R.; Valentino, M.L.; Vicenti, R.; Macciocca, M.; Cevoli, S.; Baruzzi, A.; DiMauro, S.; Carelli, V. A wide range of 3243A>G/tRNALeu(UUR) (MELAS) mutation loads may segregate in offspring through the female germline bottleneck. PLoS ONE 2014, 9, e96663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagnone, G.; Tsai, T.S.; Srirattana, K.; Rossello, F.; Powell, D.R.; Rohrer, G.; Cree, L.; Trounce, I.A.; St John, J.C. Segregation of naturally occurring mitochondrial DNA variants in a mini-pig model. Genetics 2016, 202, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, C.; Barbieri, C.; Li, M.; Bucci, L.; Monti, D.; Passarino, G.; Luiselli, D.; Franceschi, C.; Stoneking, M.; Garagnani, P. Transmission from Centenarians to their offspring of mtDNA heteroplasmy revealed by ultra-deep sequencing. Aging 2014, 6, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Zhao, X.; Li, H.; Cao, Y.; Li, W.; Ouyang, J.; Xie, L.; Liu, W. Massive parallel sequencing of mitochondrial DNA genomes from mother-child pairs using the Ion Torrent Personal Genome Machine (PGM). Forensic Sci. Int.-Genet. 2018, 32, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Schönberg, A.; Schaefer, M.; Schroeder, R.; Nasidze, I.; Stoneking, M. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 2010, 87, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.M. Best practices for reporting mtDNA heteroplasmy when using an MPS approach: Considering rates, DNA damage & drift. In Proceedings of the International Symposium on Human Identification, Minneapolis, MN, USA, September 2016. [Google Scholar]
- McElhoe, J.A.; Holland, M.M.; Makova, K.D.; Su, M.S.; Paul, I.M.; Baker, C.H.; Faith, S.A.; Young, B. Development and assessment of an optimized next-generation DNA sequencing approach for the mtgenome using the Illumina MiSeq. Forensic Sci. Int.-Genet. 2014, 13, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Schröder, R.; Ni, S.; Madea, B.; Stoneking, M. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Hayakawa, M.; Ozawa, T. Automated sequencing of mitochondrial DNA. Methods Enzymol. 1996, 264, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.M.; Pack, E.; McElhoe, J.A. Evaluation of GeneMarker® HTS for improved alignment of mtDNA MPS data, haplotype determination, and heteroplasmy assessment. Forensic Sci. Int.-Genet. 2017, 28, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.; Gusmao, L.; Hares, D.R.; Irwin, J.A.; Mayr, W.R.; Morling, N.; Pokorak, E.; Prinz, M.; Salas, A.; Schneider, P.M.; et al. DNA Commission of the International Society for Forensic Genetics: Revised and extended guidelines for mitochondrial DNA typing. Forensic Sci. Int.-Genet. 2014, 13, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Parson, W. Consistent treatment of length variants in the human mtDNA control region: A reappraisal. Int. J. Legal Med. 2008, 122, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Sawyer, S.; Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nuc. Acids Res. 2012, 40, e3. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Ijaz, U.; D’Amore, R.; Hall, N.; Sloan, W.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nuc. Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef] [PubMed]
- Bash [Unix shell program], 3.2.57(1) ed. Free Software Foundation: Boston, MA, USA, 2007.
- Team R. RStudio: Integrated Development Environment for R, 0.99.903 ed. RStudio, Inc.: Boston, MA, USA, 2015.
- Fan, L.; Yao, Y.G. An update to MitoTool: Using a new scoring system for faster mtDNA haplogroup determination. Mitochondrion 2013, 13, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Van Oven, M.; Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009, 30, E386–E394. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.; Misra, R.; Dallman, T.; Constantinidou, C.; Gharbia, S.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotech. 2012, 30, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, A.; Abeln, S.; Buijs, M.J.; Heringa, J.; Crielaard, W.; Brandt, B.W. NGS-eval: NGS Error analysis and novel sequence VAriant detection tooL. Nuc. Acids Res. 2015, 43, W301–W305. [Google Scholar] [CrossRef] [PubMed]
- Just, R.S.; Irwin, J.A.; Parson, W. DNA heteroplasmy in the emerging field of massively parallel sequencing. Forensic Sci. Int.-Genet. 2015, 18, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Stoneking, M. A New approach for detecting low-level mutations in next-generation sequence data. Genome Biol. 2012, 13, R34. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.; Russ, C.; Costello, M.; Hollinger, A.; Lennon, N.J.; Hegarty, R.; Nusbaum, C.; Jaffe, D.B. Characterizing and measuring bias in sequence data. Genome Biol. 2013, 14, R51. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nuc. Acids Res. 2008, 36, e105. [Google Scholar] [CrossRef] [PubMed]
- Meacham, F.; Boffelli, D.; Dhahbi, J.; Martin, D.I.K.; Singer, M.; Pachter, L. Identification and correction of systematic error in high-throughput sequence data. BMC Bioinform. 2011, 12, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Oshima, T.; Morimoto, T.; Ikeda, S.; Yoshikawa, H.; Shiwa, Y.; Ishikawa, S.; Linak, M.C.; Hirai, A.; Takahashi, H.; et al. Sequence-specific error profile of Illumina sequencers. Nuc. Acids Res. 2011, 39, e90. [Google Scholar] [CrossRef] [PubMed]
- Minoche, A.E.; Dohm, J.C.; Himmelbauer, H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and Genome Analyzer systems. Genome Biol. 2011, 12, R112. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mother-Child Pair | Sample Number | Shared Heteroplasmy |
|---|---|---|
| 1 | Mother-Bu (807) | A16183G (7.32%) |
| Child-Bu (803) | A16183G (6.89%) | |
| Mother-Bl (M490) | A16183G (2.81%) | |
| Child-Bl (M490-C) | A16183G (2.46%) | |
| 2 | Mother-Bu (618) | T16189C (7.74%) |
| Child-Bu (606) | T16189C (11.07%) | |
| Mother-Bl (M249) | T16189C (2.81%) | |
| Child-Bl (M249-C) | T16189C (9.92%) | |
| 3 | Mother-Bu (704) | T6152C (7.23%) |
| Child-Bu (630) | T6152C (16.37%) | |
| Mother-Bl (M234) | T6152C (5.04%) | |
| Child-Bl (M234-C) | T6152C (16.48%) | |
| 4 | Mother-Bu (762) | T10873C (2.53%) |
| Child-Bu (702) | T10873C (6.66%) | |
| Mother-Bl (M210) | ND | |
| Child-Bl (M210-C) | T10873C (5.40%) | |
| 5 | Mother-Bu (729) | A1656A (2.11%) |
| Child-Bu (684) | A1656A (2.52%) | |
| Mother-Bl (M213) | A1656A (2.77%) | |
| Child-Bl (M213-C) | A1656A (2.68%) | |
| 6 | Mother-Bu (1091) | A3243G (30.72%), A5539A (41.94%) and C16192C (19.23%) |
| Child-Bu (1111) | A3243A (33.10%), A5539G (24.54%) and C16192C (14.10%) | |
| Mother-Bl (M512) | A3243G (13.13%), A5539A (23.13%) and C16192C (22.78%) | |
| Child-Bl (M512-C) | A3243A (41.01%), A5539G (31.26%) and C16192C (17.30%) | |
| 7 | Mother-Bu (1098) | T16093C (11.53%) |
| Child-Bu (1100) | T16093T (3.45%) | |
| Mother-Bl (M520) | T16093C (9.12%) | |
| Child-Bl (M520-C) | ND | |
| 8 | Mother-Bu (1267) | T2352T (48.11%) |
| Child-Bu (1160) | T2352T (26.81%) | |
| Mother-Bl (SC16) | T2352T (47.93%) | |
| Child-Bl (SC16-C) | T2352T (26.84%) | |
| 9 | Mother-Bu (839) | C11635T (8.34%) |
| Child-Bu (1189) | C11635T (17.93%) | |
| Mother-Bl (M494) | C11635T (7.23%) | |
| Child-Bl (M494-C) | C11635T (19.88%) | |
| 10 | Mother-Bu (632) | G15047A (21.08%) |
| Child-Bu (696) | G15047A (26.67%) | |
| Mother-Bl (M236) | G15047A (19.47%) | |
| Child-Bl (M236-C) | G15047A (28.22%) | |
| 11 | Mother-Bu (531) | C5107T (9.74%) |
| Child-Bu (572) | C5107T (13.07%) | |
| Mother-Bl (M-188) | C5107T (8.19%) | |
| Child-Bl (M188-C) | C5107T (10.05%) | |
| 12 | Mother-Bu (616) | T15262C (8.36%) |
| Child-Bu (643) | T15262C (15.81%) | |
| Mother-Bl (M252) | T15262C (7.46%) | |
| Child-Bl (M252-C) | T15262C (15.49%) |
| Mother-Child Pair | Nucleotide Position | Sample Number | Major Allele | Coverage (#For:#Rev Reads) | Major Frequency (%) | Minor Allele | Coverage (#For:#Rev Reads) | Minor Frequency (%) | Gene Annotation | Synonymous (Y or N) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | T2746C | Mother - Bu (693) | T | 2920:6014 | 79.67 | C | 655:1600 | 20.11 | 16S | |
| Child - Bu (677) | T | 4838:14038 | 99.64 | C | 1:9 | 0.053 | ||||
| Mother - Bl (M207) | T | 14187:14328 | 80.3 | C | 3440:3528 | 19.62 | 16S | |||
| Child - Bl (M207-C) | T | 24044:24176 | 99.88 | C | 6:12 | 0.037 | ||||
| 2 | C16320T | Mother - Bu (406) | C | 4918:3843 | 72.33 | T | 1866:1474 | 27.57 | CR | |
| Child - Bu (444) | C | 17616:13273 | 99.92 | T | 9:7 | 0.052 | ||||
| Mother - Bl (M137) | C | 5412:4619 | 94.9 | T | 288:248 | 5.07 | CR | |||
| Child - Bl (M137-C) | C | 4232:3670 | 99.92 | T | 2:1 | 0.038 | ||||
| 3 | T9179C | Mother - Bu (1134) | T | 3063:5076 | 85.02 | C | 538:892 | 14.93 | ATP6 | N (Val to Ala) |
| Child - Bu (1099) | T | 6651:8730 | 99.82 | C | 8:7 | 0.097 | ||||
| Mother - Bl (M502G) | T | 16583:20269 | 87.14 | C | 2468:2934 | 12.77 | ATP6 | N (Val to Ala) | ||
| Child - Bl (M501) | T | 38769:44060 | 99.81 | C | 32:24 | 0.067 | ||||
| 4 | G14040A | Mother - Bu (659) | G | 5770:4227 | 92.01 | A | 474:381 | 7.86 | ND5 | Y (Gln) |
| Child - Bu (722) | G | 20789:16141 | 99.86 | A | 8:12 | 0.054 | ||||
| Mother - Bl (M242) | G | 13200:12992 | 94.07 | A | 831:811 | 5.89 | ND5 | Y (Gln) | ||
| Child - Bl (M242-C) | G | 10355:10087 | 99.88 | A | 5:5 | 0.049 | ||||
| 5 | T14461C | Mother - Bu (411) | T | 7078:7720 | 97.04 | C | 205:233 | 2.87 | ND6 | Y (Thr) |
| Child - Bu (401) | T | 16084:15992 | 99.78 | C | 35:18 | 0.165 | ||||
| Mother - Bl (M132) | T | 8475:8875 | 97.54 | C | 193:237 | 2.41 | ND6 | Y (Thr) | ||
| Child - Bl (M132-C) | T | 5854:6622 | 99.92 | C | 8:1 | 0.072 | ||||
| 6 | G11825A | Mother - Bu (711) | G | 1622:2662 | 93.41 | A | 116:184 | 6.54 | ND4 | N (Ala to Thr) |
| Child - Bu (737) | G | 8943:15813 | 99.8 | A | 8:3 | 0.044 | ||||
| Mother - Bl (M203) | G | 4728:5871 | 97.1 | A | 133:167 | 2.74 | ND4 | N (Ala to Thr) | ||
| Child - Bl (M203-C) | G | 14625:18730 | 99.88 | A | 6:5 | 0.033 | ||||
| T12375C | Mother - Bu (711) | T | 1713:1597 | 99.67 | C | 1:9 | 0.301 | |||
| Child - Bu (737) | T | 6368:6238 | 72.03 | C | 2770:2099 | 27.82 | ND5 | Y (Thr) | ||
| Mother - Bl (M203) | T | 4588:4190 | 99.66 | C | 13:12 | 0.284 | ||||
| Child - Bl (M203-C) | T | 10455:10132 | 76 | C | 3481:3008 | 23.95 | ND5 | Y (Thr) | ||
| 7 | A13790G | Mother - Bu (729) | A | 2539:943 | 99.63 | G | 1:1 | 0.057 | ||
| Child - Bu (684) | A | 5501:2427 | 88.46 | G | 650:356 | 11.22 | ND5 | N (Tyr to Cys) | ||
| Mother - Bl (M213) | A | 10487:7516 | 99.46 | G | 4:25 | 0.160 | ||||
| Child - Bl (M213-C) | A | 5900:4359 | 88.44 | G | 705:583 | 11.1 | ND5 | N (Tyr to Cys) | ||
| 8 | A200A | Mother - Bu (1098) | G | 1350:3677 | 96.58 | A | 31:139 | 3.26 | CR | |
| Child - Bu (1100) | G | 1148:3998 | 98.79 | A | 5:54 | 1.13 | ||||
| Mother - Bl (M520) | G | 821:1294 | 97.6 | A | 17:32 | 2.26 | CR | |||
| Child - Bl (M520-C) | G | 5713:9174 | 99.59 | A | 14:41 | 0.368 | ||||
| 9 | A4191T | Mother - Bu (1122) | A | 8781:10930 | 99.36 | T | 2:27 | 0.146 | ||
| Child - Bu (1119) | A | 4452:5729 | 95.5 | T | 202:243 | 4.17 | ND1 | Y (Pro) | ||
| Mother - Bl (M500) | A | 5699:6469 | 99.37 | T | 3:25 | 0.229 | ||||
| Child - Bl (M500-C) | A | 12277:14284 | 94.96 | T | 612:695 | 4.67 | ND1 | Y (Pro) | ||
| 10 | A16170G | Mother - Bu (1267) | A | 17593:21027 | 94.49 | G | 1060:1174 | 5.46 | CR | |
| Child - Bu (1160) | A | 15700:21013 | 99.95 | G | 4:5 | 0.025 | ||||
| Mother - Bl (SC16) | A | 8155:10691 | 96.19 | G | 332:413 | 3.8 | CR | |||
| Child - Bl (SC16-C) | A | 10154:13352 | 99.97 | G | 1:2 | 0.013 | ||||
| 11 | G9196A | Mother - Bu (839) | G | 6061:10535 | 97.36 | A | 172:265 | 2.56 | ATP6 | N (Asp to Asn) |
| Child - Bu (1189) | G | 4644:7284 | 99.81 | A | 0:6 | 0.050 | ||||
| Mother - Bl (M494) | G | 14294:16248 | 97.83 | A | 306:362 | 2.13 | ATP6 | N (Asp to Asn) | ||
| Child - Bl (M494-C) | G | 10236:11242 | 99.95 | A | 0:3 | 0.014 | ||||
| T3183C | Mother - Bu (839) | T | 12953:24373 | 99.67 | C | 26:53 | 0.211 | |||
| Child - Bu (1189) | T | 12210:26317 | 96.49 | C | 412:937 | 3.37 | 16S | |||
| Mother - Bl (M494) | T | 36453:46743 | 99.85 | C | 40:63 | 0.124 | ||||
| Child - Bl (M494-C) | T | 30655:39617 | 96.78 | C | 970:1333 | 3.17 | 16S | |||
| A15948G | Mother - Bu (839) | A | 15680:15099 | 99.87 | G | 13:13 | 0.084 | |||
| Child - Bu (1189) | A | 21533:19721 | 95.35 | G | 1039:902 | 4.48 | tRNAthr | |||
| Mother - Bl (M494) | A | 24430:24443 | 99.95 | G | 7:7 | 0.029 | ||||
| Child - Bl (M494-C) | A | 30887:30673 | 96.64 | G | 1074:1041 | 3.32 | tRNAthr | |||
| 12 | C11288T | Mother - Bu (740) | C | 18404:14646 | 99.97 | T | 2:1 | 0.009 | ||
| Child - Bu (718) | C | 68908:55901 | 95.69 | T | 3140:2418 | 4.26 | ND4 | Y (Leu) | ||
| Mother - Bl (M211) | C | 38204:36874 | 99.95 | T | 8:17 | 0.033 | ||||
| Child - Bl (M211-C) | C | 46511:43582 | 96.58 | T | 1651:1523 | 3.4 | ND4 | Y (Leu) | ||
| 13 | T596C | Mother - Bu (739) | T | 3088:856 | 84.7 | C | 552:155 | 15.18 | tRNAphe | |
| Child - Bu (725) | T | 9324:2728 | 99.37 | C | 3:13 | 0.132 | ||||
| Mother - Bl (M200) | T | 1745:1125 | 95.15 | C | 93:52 | 4.8 | tRNAphe | |||
| Child - Bl (M200-C) | T | 5520:3270 | 99.82 | C | 8:4 | 0.136 | ||||
| 14 | A926G | Mother - Bu (605) | A | 7528:4119 | 96.48 | G | 275:147 | 3.49 | 12S | |
| Child - Bu (619) | A | 23698:18664 | 99.95 | G | 7:5 | 0.028 | ||||
| Mother - Bl (M240) | A | 3882:3469 | 96.29 | G | 149:131 | 3.66 | 12S | |||
| Child - Bl (M240-C) | A | 4476:4483 | 99.92 | G | 2:2 | 0.043 | ||||
| 15 | A14573G | Mother - Bu (632) | A | 3406:2340 | 70.79 | G | 1390:966 | 29.02 | ND6 | N (Val to Ala) |
| Child - Bu (696) | A | 5484:4707 | 99.73 | G | 1:4 | 0.049 | ||||
| Mother - Bl (M236) | A | 7546:6525 | 77.51 | G | 2240:1839 | 22.47 | ND6 | N (Val to Ala) | ||
| Child - Bl (M236-C) | A | 8151:7438 | 99.95 | G | 3:1 | 0.026 | ||||
| A214G | Mother - Bu (632) | A | 886:2300 | 99.75 | G | 3:4 | 0.219 | |||
| Child - Bu (696) | A | 1739:2434 | 91.43 | G | 156:230 | 8.45 | CR | |||
| Mother - Bl (M236) | A | 3777:4526 | 99.99 | G | 0:1 | 0.012 | ||||
| Child - Bl (M236-C) | A | 3426:4146 | 96.87 | G | 119:124 | 3.1 | CR | |||
| 16 | A16240G | Mother - Bu (531) | A | 1424:1744 | 99.75 | G | 3:2 | 0.157 | ||
| Child - Bu (572) | A | 24590:25415 | 90.81 | G | 2527:2455 | 9.04 | CR | |||
| Mother - Bl (M-188) | A | 16073:16853 | 99.88 | G | 4:9 | 0.039 | ||||
| Child - Bl (M188-C) | A | 13732:14444 | 94.28 | G | 823:855 | 5.61 | CR | |||
| 17 | A9983G | Mother - Bu (616) | A | 2944:7014 | 99.83 | G | 5:12 | 0.170 | ||
| Child - Bu (643) | A | 11623:20991 | 97.33 | G | 314:547 | 2.56 | CO3 | Y (Trp) | ||
| Mother - Bl (M252) | A | 11354:14962 | 99.83 | G | 18:22 | 0.152 | ||||
| Child - Bl (M252-C) | A | 17123:22272 | 97.91 | G | 346:468 | 2.02 | CO3 | Y (Trp) |
| rCRS>A | rCRS>C | rCRS>G | rCRS>T | |
|---|---|---|---|---|
| Error Rate | 0.0343 | 0.0565 | 0.0331 | 0.0304 |
| rCRS>A error | |||
|---|---|---|---|
| Adjacent Sequence | rCRS nt | np | Error |
| AAA*TAC | C | 2785 | 0.669 |
| TCA*AAG | T | 2445 | 0.722 |
| ATA*AAAA | T | 6415 | 0.732 |
| AAA*AGT | C | 2756 | 0.836 |
| AGA*GAG | C | 2718 | 0.875 |
| AAG*AAC | G | 2471 | 0.900 |
| CAA*ACG | G | 2716 | 0.920 |
| GAA*ACC | G | 2724 | 1.013 |
| AAC*AAC | T | 10,304 | 1.018 |
| AAAAA*AAAAAA | T | 8496 | 1.079 |
| AAC*AAAA | C | 8523 | 1.095 |
| CTA*AAA | G | 10,260 | 1.130 |
| AAG*AAA | G | 2449 | 1.223 |
| CCA*AAAAA | T | 8490 | 1.395 |
| CTA*AAA | C | 10,296 | 1.421 |
| AGT*AAA | T | 1115 | 1.517 |
| CAC*AAA | C | 3477 | 1.668 |
| TAA*ACA | C | 2708 | 2.048 |
| AAC*AAAA | G | 8533 | 2.355 |
| GGT*AAAAAAA | T | 2456 | 2.457 |
| CCA*AAAA | T | 3464 | 2.784 |
| CGG*AAA | C | 2479 | 3.371 |
| AAAAA*TTC | T | 2806 | 3.634 |
| CCCC*AAAA | T | 3488 | 4.629 |
| AAAAAAAG*AAAAGG | T | 2465 | 4.787 |
| rCRS>C error | |||
| Adjacent Sequence | rCRS nt | np | Error |
| AAC*CTA | A | 1142 | 1.971 |
| TCT*CAC | T | 3473 | 1.996 |
| CTC*CCA | A | 14,914 | 2.010 |
| ATT*CCC | A | 2412 | 2.051 |
| TAG*CCT | G | 8573 | 2.157 |
| ATC*CCG | A | 3523 | 2.214 |
| TCC*CCA | A | 297 | 2.261 |
| AAC*CGG | T | 2475 | 2.363 |
| CCCC*CCCC | A | 14,813 | 2.532 |
| TAA*CCT | A | 1104 | 2.629 |
| CCCCCCC*CCCCC | T | 310 | 2.702 |
| TAC*CCT | A | 6355 | 2.840 |
| TGA*CCC | G | 10,290 | 3.220 |
| CAA*CCCCCCC | A | 302 | 3.259 |
| GAT*CCCC | A | 1082 | 3.365 |
| TCT*CCA | A | 3505 | 3.387 |
| CAA*CCC | A | 8512 | 3.419 |
| ATT*CCT | A | 10,239 | 3.531 |
| CCC*CCC | A | 6316 | 3.635 |
| AAA*CTC | A | 3468 | 4.003 |
| TCC*CCC | A | 5208 | 4.003 |
| CCT*CCC | A | 8577 | 4.318 |
| AGA*CCCC | G | 3483 | 4.614 |
| ACA*CCC | A | 3447 | 4.753 |
| CAA*CCT | T | 8567 | 4.799 |
| TTC*CCA | A | 3475 | 4.954 |
| CCT*CCA | A | 10,283 | 5.115 |
| AAA*CCC | A | 3492 | 5.852 |
| ATT*CCCCC | G | 8557 | 5.990 |
| AAC*CCC | A | 5192 | 6.157 |
| CTA*CCT | A | 10,306 | 6.723 |
| TTTT*CCCC | A | 10,277 | 7.210 |
| AAA*CCCCC | A | 6419 | 10.035 |
| ATC*CCC | A | 3511 | 11.371 |
| CTT*CCCC | A | 2487 | 14.821 |
| rCRS>G error | |||
| Adjacent Sequence | rCRS nt | np | Error |
| CAC*CCC | T | 466 | 0.301 |
| CAG*GCC | A | 3243 | 0.301 |
| AAC*GGC | T | 5717 | 0.311 |
| GAG*GTT | T | 944 | 0.320 |
| GGGG*AGC | A | 16,037 | 0.325 |
| AAA*CCCCC | A | 16,183 | 0.326 |
| CCC*CCC | A | 16,293 | 0.328 |
| CAG*TTA | T | 578 | 0.329 |
| TGG*GAT | T | 2010 | 0.329 |
| AAC*CGG | T | 2475 | 0.337 |
| CGC*GAC | T | 3456 | 0.347 |
| TTA*CCC | C | 10,287 | 0.358 |
| AGG*GTA | T | 1335 | 0.376 |
| AAG*GCC | A | 5539 | 0.382 |
| AGG*GGC | T | 1349 | 0.406 |
| CCG*ATA | T | 7429 | 0.444 |
| CGG*GCT | T | 1180 | 0.487 |
| GGG*ATA | C | 2703 | 0.501 |
| AAC*GCT | T | 1129 | 0.577 |
| AAC*GGG | T | 1071 | 0.628 |
| AAG*GCC | A | 3482 | 0.672 |
| AGG*CCT | T | 2778 | 0.950 |
| TGG*TTC | T | 7480 | 1.069 |
| AGG*TAT | T | 7522 | 1.183 |
| TGG*GCT | A | 2734 | 2.458 |
| rCRS>T error | |||
| Adjacent Sequence | rCRS nt | np | Error |
| ATC*CCC | A | 16 | 0.345 |
| CAA*CCCCCCC | A | 302 | 0.343 |
| GTC*CCCCCC | A | 432 | 0.322 |
| ACA*TTA | G | 1113 | 0.320 |
| GAT*AAAA | T | 2352 | 0.834 |
| AAG*TTA | G | 2454 | 0.630 |
| TTT*ATT | A | 2740 | 1.058 |
| TTT*TTA | A | 2745 | 0.679 |
| ATT*ATG | A | 2748 | 0.570 |
| TAT*CCC | A | 4455 | 0.347 |
| CTA*TAC | C | 5107 | 0.360 |
| TCT*CCT | A | 5347 | 0.348 |
| ATC*CCT | A | 7649 | 0.346 |
| TCT*TTC | G | 8541 | 0.509 |
| TCG*TTC | C | 8546 | 0.579 |
| TTC*TTC | A | 8550 | 0.382 |
| AGC*GGC | G | 8856 | 0.410 |
| ACC*CCT | A | 9425 | 0.333 |
| CAG*CAC | C | 11,635 | 0.377 |
| CCCCCC*CTA | A | 11,873 | 0.354 |
| ACC*CCC | A | 12,400 | 0.331 |
| GCT*CCT | A | 14,988 | 0.370 |
| ATC*CCT | A | 15,401 | 0.426 |
| TCC*CCC | A | 15,408 | 0.579 |
| CCC*CCC | A | 16,293 | 0.341 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holland, M.M.; Makova, K.D.; McElhoe, J.A. Deep-Coverage MPS Analysis of Heteroplasmic Variants within the mtGenome Allows for Frequent Differentiation of Maternal Relatives. Genes 2018, 9, 124. https://doi.org/10.3390/genes9030124
Holland MM, Makova KD, McElhoe JA. Deep-Coverage MPS Analysis of Heteroplasmic Variants within the mtGenome Allows for Frequent Differentiation of Maternal Relatives. Genes. 2018; 9(3):124. https://doi.org/10.3390/genes9030124
Chicago/Turabian StyleHolland, Mitchell M., Kateryna D. Makova, and Jennifer A. McElhoe. 2018. "Deep-Coverage MPS Analysis of Heteroplasmic Variants within the mtGenome Allows for Frequent Differentiation of Maternal Relatives" Genes 9, no. 3: 124. https://doi.org/10.3390/genes9030124
APA StyleHolland, M. M., Makova, K. D., & McElhoe, J. A. (2018). Deep-Coverage MPS Analysis of Heteroplasmic Variants within the mtGenome Allows for Frequent Differentiation of Maternal Relatives. Genes, 9(3), 124. https://doi.org/10.3390/genes9030124