Re-Arrangements in the Cytoplasmic Distribution of Small RNAs Following the Maternal-to-Zygotic Transition in Drosophila Embryos



Abstract
:1. Introduction
2. Materials and Methods
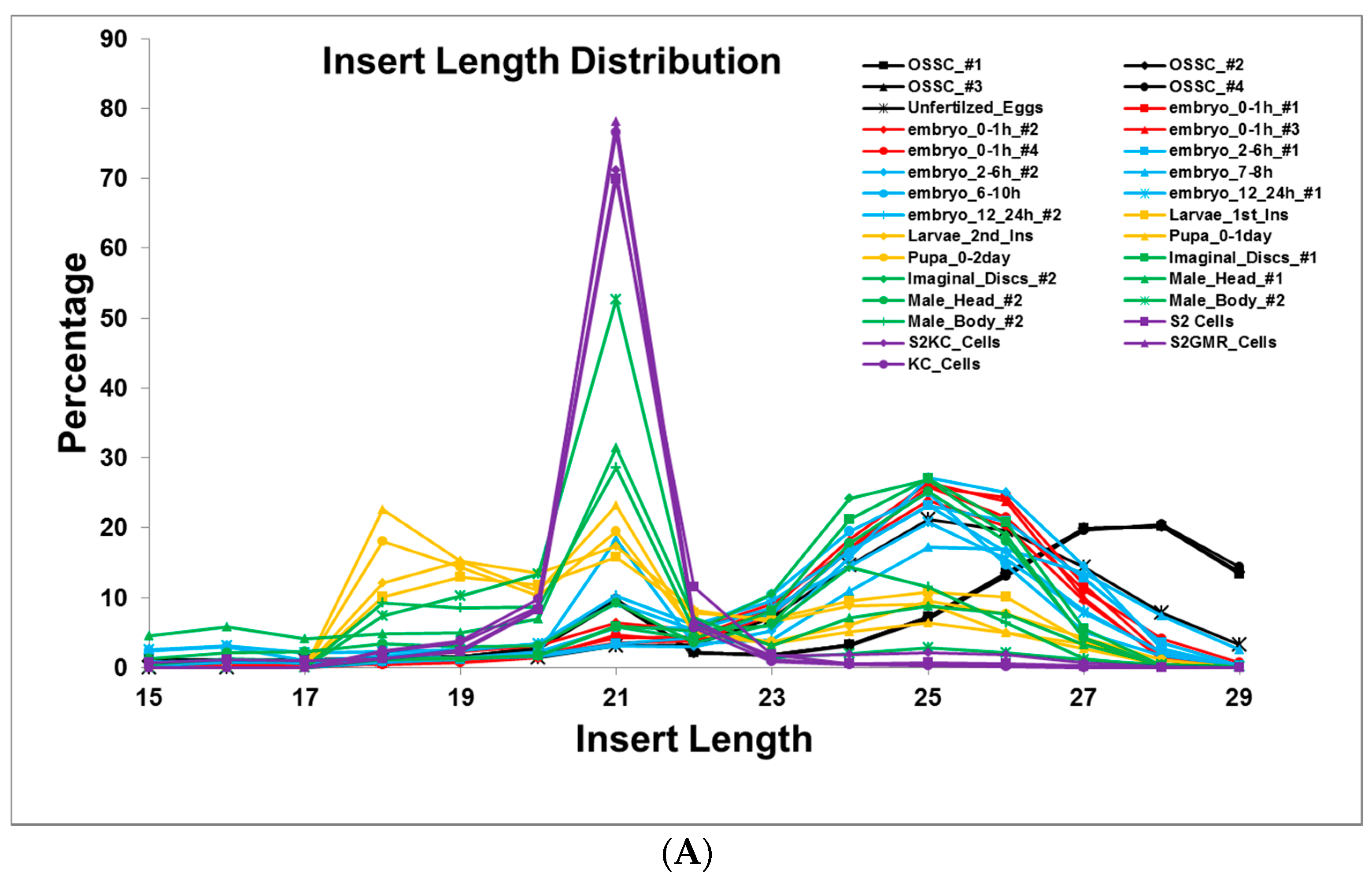
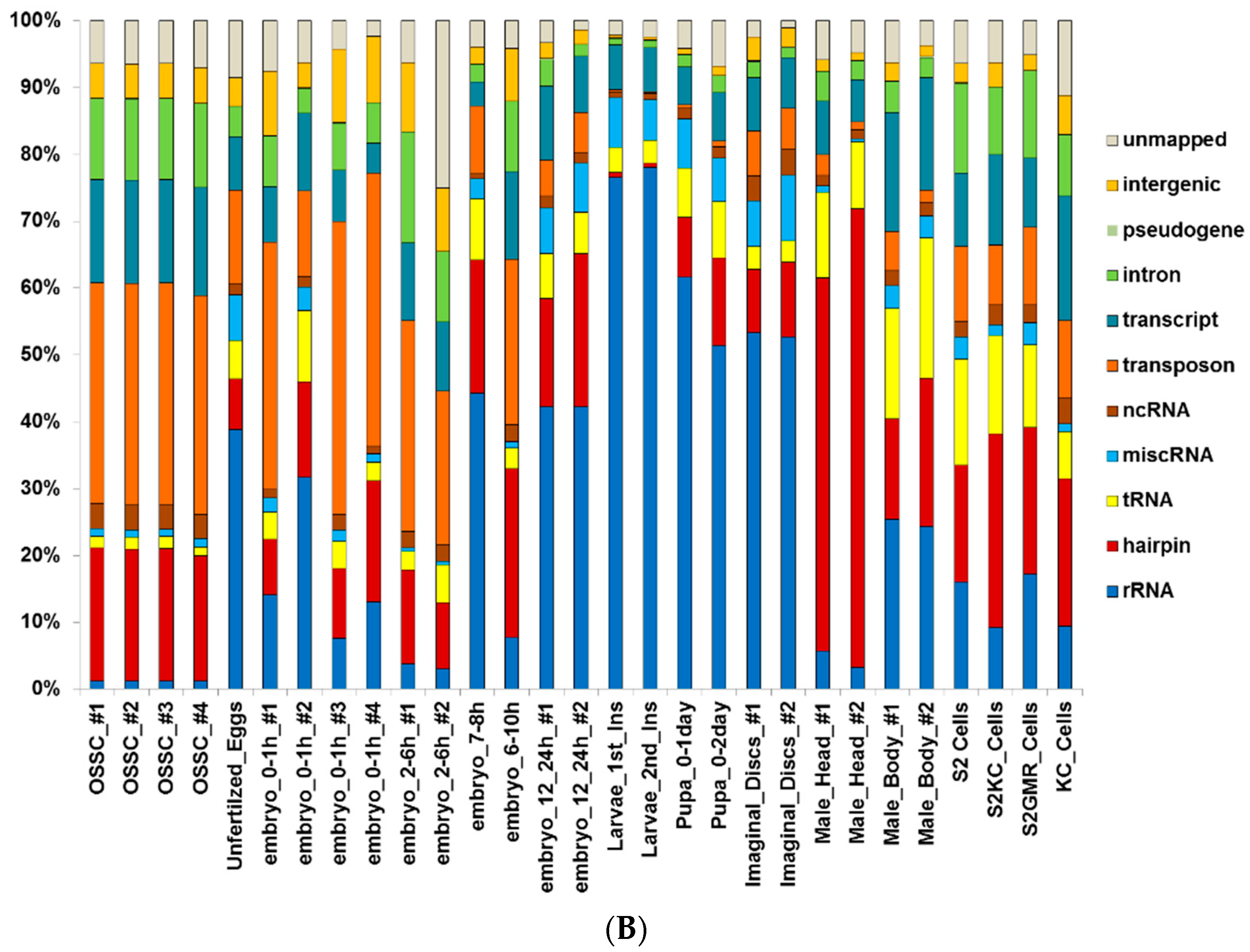
2.1. Small RNA Deep-Sequencing Data Analysis
2.2. Quantitative PCR Analysis
3. Results
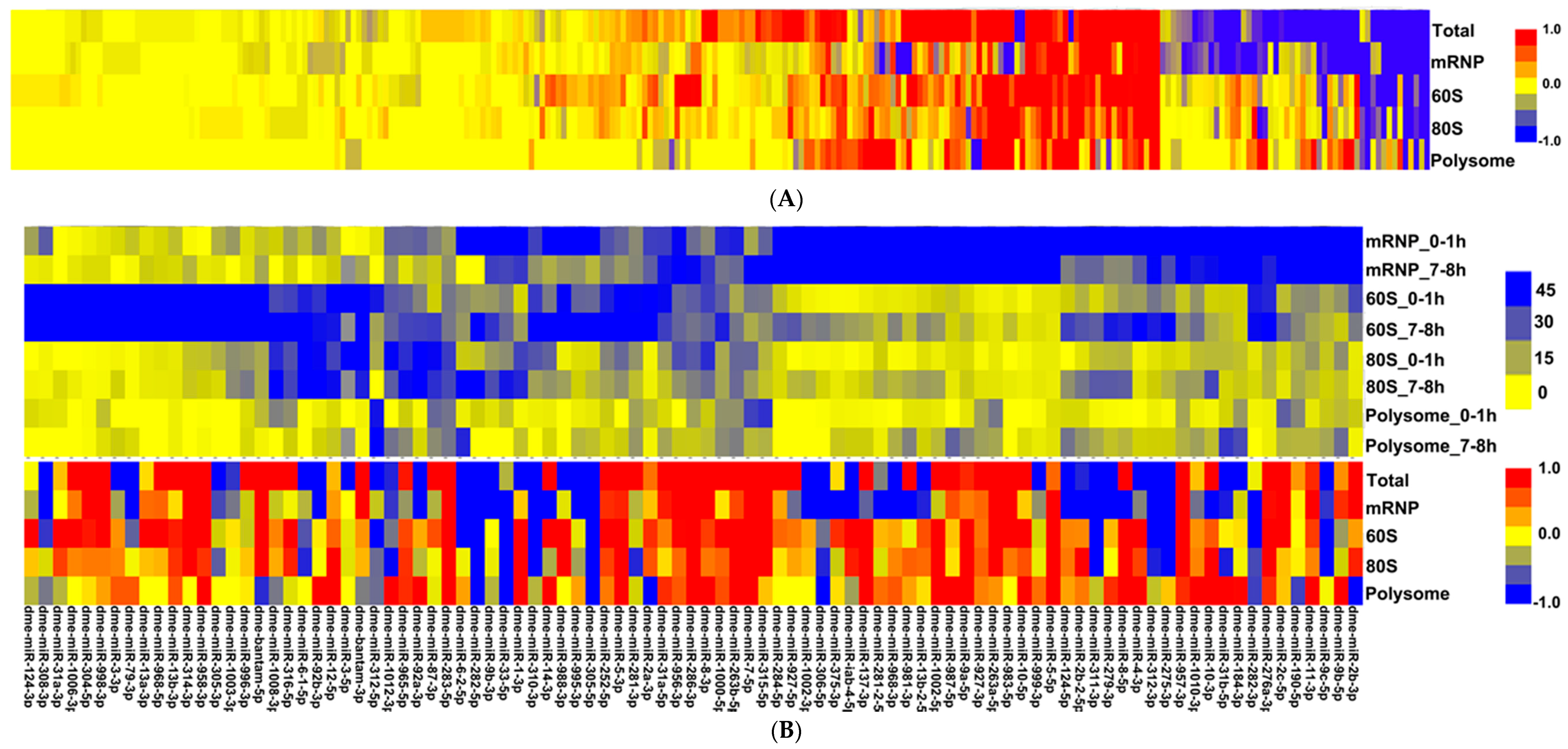
3.1. Small RNA Populations Localize in Distinct Cytoplasmic Reservoirs
3.2. Micro RNAs Interact with Cellular Translational Machinery at All States
3.3. Micro RNA Cluster Behaviours and miRNA Editing
3.4. Cytoplasmic Micro RNA Re-Arrangement Does Not Correlate with Target Abundance
3.5. Transposon-Derived Small Interfering RNAs and PIWI-Interacting RNAs Interact with Different Complexes
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Renzis, S.; Elemento, O.; Tavazoie, S.; Wieschaus, E.F. Unmasking activation of the zygotic genome using chromosomal deletions in the Drosophila embryo. PLoS Biol. 2007, 5, e117. [Google Scholar] [CrossRef]
- Tadros, W.; Goldman, A.L.; Babak, T.; Menzies, F.; Vardy, L.; Orr-Weaver, T.; Hughes, T.R.; Westwood, J.T.; Smibert, C.A.; Lipshitz, H.D. Smaug is a major regulator of maternal mRNA destabilization in Drosophila and its translation is activated by the PAN GU kinase. Dev. Cell 2007, 12, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C.; Cochella, L. A framework for understanding the roles of miRNAs in animal development. Development 2017, 144, 2548–2559. [Google Scholar] [CrossRef] [PubMed]
- Bourc’his, D.; Voinnet, O. A small-RNA perspective on gametogenesis, fertilization, and early zygotic development. Science 2010, 330, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Bushati, N.; Stark, A.; Brennecke, J.; Cohen, S.M. Temporal reciprocity of miRNAs and their targets during the maternal-to-zygotic transition in Drosophila. Curr. Biol. 2008, 18, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Flemr, M.; Stein, P.; Berninger, P.; Malik, R.; Zavolan, M.; Svoboda, P.; Schultz, R.M. MicroRNA activity is suppressed in mouse oocytes. Curr. Biol. 2010, 20, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Rouget, C.; Papin, C.; Boureux, A.; Meunier, A.C.; Franco, B.; Robine, N.; Lai, E.C.; Pelisson, A.; Simonelig, M. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature 2010, 467, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Weick, E.M.; Miska, E.A. PiRNAs: From biogenesis to function. Development 2014, 141, 3458–3471. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Finn, K.J.; Ji, X.; Baillat, D.; Gregory, R.I.; Liebhaber, S.A.; Pasquinelli, A.E.; Shiekhattar, R. MicroRNA silencing through risc recruitment of Eif6. Nature 2007, 447, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiriakidou, M.; Tan, G.S.; Lamprinaki, S.; De Planell-Saguer, M.; Nelson, P.T.; Mourelatos, Z. An mRNA m7G cap binding-like motif within human Ago2 represses translation. Cell 2007, 129, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.P.; Bordeleau, M.E.; Pelletier, J.; Sharp, P.A. Short RNAs repress translation after initiation in mammalian cells. Mol. Cell 2006, 21, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.A.; Lagos-Quintana, M.; Yalcin, A.; Zavolan, M.; Marks, D.; Snyder, B.; Gaasterland, T.; Meyer, J.; Tuschl, T. The small RNA profile during Drosophila melanogaster development. Dev. Cell 2003, 5, 337–350. [Google Scholar] [CrossRef]
- Okamura, K.; Lai, E.C. Endogenous small interfering RNAs in animals. Nat. Rev. Mol. Cell Biol. 2008, 9, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Barckmann, B.; Pierson, S.; Dufourt, J.; Papin, C.; Armenise, C.; Port, F.; Grentzinger, T.; Chambeyron, S.; Baronian, G.; Desvignes, J.P.; et al. Aubergine iCLIP reveals piRNA-dependent decay of mRNAs involved in germ cell development in the early embryo. Cell Rep. 2015, 12, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, J.M. Ancient endo-siRNA pathways reveal new tricks. Curr. Biol. 2014, 24, R703–R715. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Schuster, A.; Tang, C.; Yu, T.; Ortogero, N.; Bao, J.; Zheng, H.; Yan, W. Sperm-borne miRNAs and endo-siRNAs are important for fertilization and preimplantation embryonic development. Development 2016, 143, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, J.; Tai, Y.; Zhang, X.; Zhang, J.; Liu, S.; Lv, J.; Liu, Z.; Kong, Q. Identification and characterization of l1-specific endo-siRNAs essential for early embryonic development in pig. Oncotarget 2017, 8, 23167–23176. [Google Scholar] [CrossRef] [PubMed]
- Aboobaker, A.A.; Tomancak, P.; Patel, N.; Rubin, G.M.; Lai, E.C. Drosophila microRNAs exhibit diverse spatial expression patterns during embryonic development. Proc. Natl. Acad. Sci. USA 2005, 102, 18017–18022. [Google Scholar] [CrossRef] [PubMed]
- Leaman, D.; Chen, P.Y.; Fak, J.; Yalcin, A.; Pearce, M.; Unnerstall, U.; Marks, D.S.; Sander, C.; Tuschl, T.; Gaul, U. Antisense-mediated depletion reveals essential and specific functions of microRNAs in Drosophila development. Cell 2005, 121, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Song, S.; Weng, R.; Verma, P.; Kugler, J.M.; Buescher, M.; Rouam, S.; Cohen, S.M. Systematic study of Drosophila microRNA functions using a collection of targeted knockout mutations. Dev. Cell 2014, 31, 784–800. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Kaneda, M.; O’Carroll, D.; Hajkova, P.; Barton, S.C.; Sun, Y.A.; Lee, C.; Tarakhovsky, A.; Lao, K.; Surani, M.A. Maternal microRNAs are essential for mouse zygotic development. Genes Dev. 2007, 21, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.; Choudhary, A.; Patowary, A.; Singh, A.R.; Bhatia, S.; Sivasubbu, S.; Chandrasekaran, S.; Pillai, B. Mir-34 is maternally inherited in Drosophila melanogaster and Danio rerio. Nucleic Acids Res. 2013, 41, 4470–4480. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, P.; Flemr, M. The role of miRNAs and endogenous siRNAs in maternal-to-zygotic reprogramming and the establishment of pluripotency. EMBO Rep. 2010, 11, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Nien, C.Y.; Liang, H.L.; Rushlow, C. Co-activation of microRNAs by Zelda is essential for early Drosophila development. Development 2014, 141, 2108–2118. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Choi, Y.; Kim, K.; Jin, H.; Lim, J.; Nguyen, T.A.; Yang, J.; Jeong, M.; Giraldez, A.J.; Yang, H.; et al. Adenylation of maternally inherited microRNAs by Wispy. Mol. Cell 2014, 56, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Lin, H. Posttranscriptional regulation of gene expression by PIWI proteins and piRNAs. Mol. Cell 2014, 56, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Valencia-Sanchez, M.A.; Hannon, G.J.; Parker, R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat. Cell Biol. 2005, 7, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Maroney, P.A.; Yu, Y.; Fisher, J.; Nilsen, T.W. Evidence that microRNAs are associated with translating messenger RNAs in human cells. Nat. Struct. Mol. Biol. 2006, 13, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Molotski, N.; Soen, Y. Differential association of microRNAs with polysomes reflects distinct strengths of interactions with their mRNA targets. RNA 2012, 18, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.H.; Isaji, M.; Carthew, R.W. Functionally diverse microRNA effector complexes are regulated by extracellular signaling. Mol. Cell 2013, 52, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Goktas, C.; Yigit, H.; Cosacak, M.I.; Akgul, B. Differentially expressed tRNA-derived small RNAs co-sediment primarily with non-polysomal fractions in Drosophila. Genes 2017, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- De Hoon, M.J.; Taft, R.J.; Hashimoto, T.; Kanamori-Katayama, M.; Kawaji, H.; Kawano, M.; Kishima, M.; Lassmann, T.; Faulkner, G.J.; Mattick, J.S.; et al. Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res. 2010, 20, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Gramates, L.S.; Marygold, S.J.; Santos, G.D.; Urbano, J.M.; Antonazzo, G.; Matthews, B.B.; Rey, A.J.; Tabone, C.J.; Crosby, M.A.; Emmert, D.B.; et al. Flybase at 25: Looking to the future. Nucleic Acids Res. 2017, 45, D663–D671. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. Genbank. Nucleic Acids Res 2008, 36, D25–D30. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.; Aravin, A.A.; Stark, A.; Dus, M.; Kellis, M.; Sachidanandam, R.; Hannon, G.J. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 2007, 128, 1089–1103. [Google Scholar] [CrossRef] [PubMed]
- De Hoon, M.J.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, A.J. Java Treeview—Extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef] [PubMed]
- Celniker, S.E.; Dillon, L.A.; Gerstein, M.B.; Gunsalus, K.C.; Henikoff, S.; Karpen, G.H.; Kellis, M.; Lai, E.C.; Lieb, J.D.; MacAlpine, D.M.; et al. Unlocking the secrets of the genome. Nature 2009, 459, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Bor, Y.C.; Swartz, J.; Li, Y.; Coyle, J.; Rekosh, D.; Hammarskjold, M.-L. Northern blot analysis of mRNA from mammalian polyribosomes. Protocol Exchange 2006. [Google Scholar] [CrossRef]
- Altuvia, Y.; Landgraf, P.; Lithwick, G.; Elefant, N.; Pfeffer, S.; Aravin, A.; Brownstein, M.J.; Tuschl, T.; Margalit, H. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005, 33, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Baskerville, S.; Bartel, D.P. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 2005, 11, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Haubrock, M.; Cao, K.M.; Hua, X.; Zhang, C.Y.; Wingender, E.; Li, J. Regulatory coordination of clustered microRNAs based on microRNA-transcription factor regulatory network. BMC Syst. Biol. 2011, 5, 199. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Grivna, S.T.; Pyhtila, B.; Lin, H. MIWI associates with translational machinery and PIWI-interacting RNAs (piRNAs) in regulating spermatogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 13415–13420. [Google Scholar] [CrossRef] [PubMed]
- Ghildiyal, M.; Seitz, H.; Horwich, M.D.; Li, C.; Du, T.; Lee, S.; Xu, J.; Kittler, E.L.; Zapp, M.L.; Weng, Z.; et al. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science 2008, 320, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.; Gaidatzis, D.; Pfeffer, S.; Lagos-Quintana, M.; Landgraf, P.; Iovino, N.; Morris, P.; Brownstein, M.J.; Kuramochi-Miyagawa, S.; Nakano, T.; et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 2006, 442, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Klattenhoff, C.; Xi, H.; Li, C.; Lee, S.; Xu, J.; Khurana, J.S.; Zhang, F.; Schultz, N.; Koppetsch, B.S.; Nowosielska, A.; et al. The Drosophila Hp1 homolog rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell 2009, 138, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.J.; Okamura, K.; Martin, R.; Lai, E.C. Endogenous RNA interference provides a somatic defense against Drosophila transposons. Curr. Biol. 2008, 18, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Giraldez, A.J.; Mishima, Y.; Rihel, J.; Grocock, R.J.; Van Dongen, S.; Inoue, K.; Enright, A.J.; Schier, A.F. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science 2006, 312, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Tchurikov, N.A.; Kretova, O.V. Both piRNA and siRNA pathways are silencing transcripts of the suffix element in the Drosophila melanogaster germline and somatic cells. PLoS ONE 2011, 6, e21882. [Google Scholar] [CrossRef] [PubMed]
- Zong, Q.; Schummer, M.; Hood, L.; Morris, D.R. Messenger RNA translation state: The second dimension of high-throughput expression screening. Proc. Natl. Acad. Sci. USA 1999, 96, 10632–10636. [Google Scholar] [CrossRef] [PubMed]
- Krichevsky, A.M.; King, K.S.; Donahue, C.P.; Khrapko, K.; Kosik, K.S. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 2003, 9, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Suh, N.; Baehner, L.; Moltzahn, F.; Melton, C.; Shenoy, A.; Chen, J.; Blelloch, R. MicroRNA function is globally suppressed in mouse oocytes and early embryos. Curr. Biol. 2010, 20, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Dallaire, A.; Simard, M.J. The implication of microRNAs and endo-siRNAs in animal germline and early development. Dev. Biol. 2016, 416, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Suh, N.; Blelloch, R. Small RNAs in early mammalian development: From gametes to gastrulation. Development 2011, 138, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Fagegaltier, D.; Bouge, A.L.; Berry, B.; Poisot, E.; Sismeiro, O.; Coppee, J.Y.; Theodore, L.; Voinnet, O.; Antoniewski, C. The endogenous siRNA pathway is involved in heterochromatin formation in Drosophila. Proc. Natl. Acad. Sci. USA 2009, 106, 21258–21263. [Google Scholar] [CrossRef] [PubMed]
- Karaiskos, S.; Naqvi, A.S.; Swanson, K.E.; Grigoriev, A. Age-driven modulation of tRNA-derived fragments in Drosophila and their potential targets. Biol. Direct. 2015, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Sobala, A.; Hutvagner, G. Small RNAs derived from the 5’ end of tRNA can inhibit protein translation in human cells. RNA Biol. 2013, 10, 553–563. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hairpin | Mature_3bp | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mRNP | 60S | 80S | Polysome | Total | mRNP | 60S | 80S | Polysome | Total | |||||||||||
| 0–1 h | 7–8 h | 0–1 h | 7–8 h | 0–1 h | 7–8 h | 0–1 h | 7–8 h | 0–1 h | 7–8 h | 0–1 h | 7–8 h | 0–1 h | 7–8 h | 0–1 h | 7–8 h | 0–1 h | 7–8 h | 0–1 h | 7–8 h | |
| dme-bantam-3p | 0.3 | 3.4 | 36.3 | 27.3 | 35.9 | 26.4 | 2.9 | 1.0 | 1.5 | 30.3 | 0.3 | 3.3 | 35.7 | 26.2 | 35.7 | 26.1 | 2.6 | 0.9 | 1.5 | 30.1 |
| dme-miR-1-3p | 7.7 | 5.5 | 3.1 | 7.9 | 14.8 | 19.9 | 1.0 | 7.5 | 43.9 | 13.7 | 7.7 | 5.5 | 3.1 | 7.9 | 14.8 | 19.9 | 1.0 | 7.4 | 43.9 | 13.7 |
| dme-miR-184-3p | 17.5 | 9.6 | 7.6 | 4.6 | 10.4 | 6.1 | 27.0 | 25.9 | 11.1 | 3.6 | 17.5 | 9.6 | 7.6 | 4.6 | 10.4 | 6.1 | 27.0 | 25.9 | 11.1 | 3.6 |
| dme-miR-263a-5p | 18.0 | 24.7 | 1.5 | 4.2 | 3.4 | 5.1 | 33.1 | 22.2 | 3.5 | 9.0 | 18.0 | 24.7 | 1.5 | 4.2 | 3.4 | 5.1 | 33.1 | 22.1 | 3.5 | 9.0 |
| dme-miR-283-5p | 0.2 | 0.3 | 0.9 | 1.3 | 1.3 | 1.9 | 1.4 | 1.8 | 0.2 | 1.4 | 0.2 | 0.3 | 0.9 | 1.2 | 1.3 | 1.8 | 1.4 | 1.8 | 0.1 | 1.4 |
| dme-miR-286-3p | 2.0 | 10.8 | 7.1 | 18.1 | 7.5 | 18.9 | 6.8 | 10.3 | 2.2 | 11.6 | 2.0 | 10.8 | 7.1 | 18.1 | 7.5 | 18.9 | 6.8 | 10.3 | 2.2 | 11.5 |
| dme-miR-305-5p | 1.0 | 0.0 | 1.5 | 0.3 | 0.4 | 0.1 | 0.3 | 0.0 | 1.7 | 0.1 | 1.0 | 0.0 | 1.3 | 0.2 | 0.3 | 0.0 | 0.3 | 0.0 | 1.7 | 0.1 |
| dme-miR-306-5p | 2.7 | 0.0 | 0.1 | 0.0 | 0.1 | 0.0 | 0.1 | 0.0 | 2.4 | 0.1 | 2.7 | 0.0 | 0.1 | 0.0 | 0.1 | 0.0 | 0.1 | 0.0 | 2.4 | 0.1 |
| dme-miR-311-3p | 2.1 | 0.0 | 0.6 | 0.1 | 1.0 | 0.1 | 0.2 | 0.1 | 2.6 | 0.1 | 2.1 | 0.0 | 0.6 | 0.1 | 1.0 | 0.1 | 0.2 | 0.1 | 2.6 | 0.1 |
| dme-miR-315-5p | 0.2 | 0.8 | 0.6 | 0.8 | 0.3 | 0.5 | 1.4 | 0.8 | 0.2 | 2.0 | 0.2 | 0.8 | 0.6 | 0.8 | 0.3 | 0.5 | 1.4 | 0.8 | 0.2 | 2.0 |
| dme-miR-5-5p | 16.4 | 27.2 | 3.4 | 4.3 | 2.4 | 3.5 | 5.9 | 7.0 | 3.3 | 4.5 | 16.4 | 27.2 | 3.1 | 3.9 | 2.3 | 3.3 | 5.9 | 6.9 | 3.2 | 4.2 |
| dme-miR-7-5p | 0.1 | 1.5 | 0.5 | 1.2 | 0.6 | 1.6 | 1.1 | 1.4 | 0.3 | 1.0 | 0.1 | 1.5 | 0.5 | 1.2 | 0.6 | 1.6 | 1.1 | 1.4 | 0.3 | 1.0 |
| dme-miR-8-3p | 0.9 | 1.2 | 3.0 | 2.9 | 3.3 | 3.0 | 1.2 | 1.7 | 1.3 | 2.5 | 0.5 | 1.1 | 2.7 | 2.5 | 3.1 | 2.7 | 1.2 | 1.6 | 1.2 | 2.2 |
| dme-miR-92b-3p | 0.3 | 0.0 | 1.6 | 1.0 | 2.0 | 1.2 | 0.2 | 0.3 | 0.8 | 0.1 | 0.3 | 0.0 | 1.6 | 0.9 | 2.0 | 1.2 | 0.2 | 0.3 | 0.8 | 0.1 |
| dme-miR-958-3p | 0.0 | 0.1 | 4.2 | 5.1 | 0.5 | 0.6 | 0.1 | 0.2 | 0.0 | 0.8 | 0.0 | 0.1 | 4.1 | 5.0 | 0.5 | 0.6 | 0.0 | 0.1 | 0.0 | 0.8 |
| dme-miR-9a-5p | 6.0 | 5.3 | 1.6 | 3.5 | 1.7 | 2.9 | 1.7 | 8.6 | 4.1 | 6.4 | 6.0 | 5.3 | 1.6 | 3.4 | 1.7 | 2.9 | 1.7 | 8.6 | 4.1 | 6.4 |
| dme-miR-9c-5p | 11.5 | 4.1 | 8.8 | 2.4 | 6.0 | 1.8 | 9.1 | 4.4 | 9.4 | 2.6 | 11.5 | 4.1 | 8.8 | 2.4 | 6.0 | 1.8 | 9.0 | 4.4 | 9.4 | 2.5 |
| dme-miR-iab-4-5p | 1.2 | 0.1 | 0.1 | 0.1 | 0.0 | 0.0 | 0.1 | 0.0 | 0.5 | 0.1 | 1.6 | 0.1 | 0.0 | 0.1 | 0.0 | 0.0 | 0.2 | 0.1 | 0.7 | 0.1 |
| G | miRNA | Tot | mRNP | 60S | 80S | Poly | G | miRNA | Tot | mRNP | 60S | 80S | Poly |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | dme-miR-286-3p | 2.6 | 3.1 | 2.2 | 2.1 | 1.6 | 2 | dme-miR-11-3p | 0.8 | 1.3 | 0.8 | 0.5 | 2.4 |
| 1 | dme-miR-283-5p | 3.6 | 1.5 | 1.3 | 1.3 | 1.3 | 2 | dme-miR-9b-5p | 0.4 | 0.8 | −0.4 | −0.2 | 1.7 |
| 1 | dme-miR-7-5p | 2.0 | 4.5 | 2.0 | 2.1 | 1.3 | 2 | dme-miR-1012-3p | −0.8 | −0.8 | −0.5 | −1.0 | 1.4 |
| 1 | dme-miR-957-3p | 2.6 | 1.3 | 1.2 | 1.2 | 0.8 | 2 | dme-miR-1010-3p | 0.2 | −0.6 | 0.4 | 0.5 | 1.4 |
| 1 | dme-miR-263a-5p | 1.6 | 1.1 | 2.3 | 1.3 | 0.4 | 2 | dme-miR-5-5p | 0.6 | 1.4 | 1.2 | 1.3 | 1.2 |
| 1 | dme-miR-315-5p | 3.9 | 2.6 | 1.3 | 1.3 | 0.2 | 2 | dme-miR-12-5p | 0.3 | 0.4 | 0.6 | 1.2 | 1.1 |
| 1 | dme-miR-314-3p | 5.6 | 2.1 | 1.3 | 1.0 | 0.0 | 2 | dme-miR-13a-3p | 0.2 | 0.6 | 1.3 | 0.2 | 0.0 |
| 1 | dme-bantam-5p | 2.6 | 1.7 | 1.5 | 1.2 | −0.8 | 2 | dme-miR-31a-3p | 0.2 | 0.0 | 1.0 | 0.7 | −0.3 |
| 1 | dme-miR-958-3p | 7.0 | 3.6 | 1.1 | 0.8 | 1.8 | 2 | dme-miR-124-3p | 0.0 | −0.3 | 1.2 | 0.2 | −0.3 |
| 1 | dme-miR-956-3p | 3.3 | 1.2 | 1.0 | 0.6 | 0.7 | 2 | dme-miR-965-5p | 0.9 | −0.4 | 0.7 | 0.8 | 0.8 |
| 1 | dme-miR-998-3p | 1.3 | 1.2 | 1.5 | 0.5 | 0.4 | 2 | dme-miR-1003-3p | −0.8 | −0.5 | 0.1 | 0.0 | 0.0 |
| 1 | dme-miR-2c-5p | 1.4 | 1.2 | 1.6 | 0.2 | 0.2 | 2 | dme-miR-190-5p | 0.4 | 0.1 | 0.0 | 0.1 | −0.7 |
| 1 | dme-miR-304-5p | 6.0 | 1.5 | 0.9 | 0.5 | 0.1 | 2 | dme-miR-282-3p | −0.1 | −1.9 | −1.7 | −0.7 | −0.7 |
| 1 | dme-miR-8-3p | 1.1 | 1.8 | 0.8 | 0.5 | 1.3 | 2 | dme-miR-2a-3p | 0.6 | 0.2 | 0.8 | 0.5 | −1.5 |
| 1 | dme-miR-983-5p | 2.0 | 1.2 | 0.7 | 0.3 | 0.0 | 2 | dme-miR-33-5p | −0.3 | −2.4 | −1.6 | −1.1 | −1.9 |
| 1 | dme-bantam-3p | 4.6 | 4.3 | 0.4 | 0.3 | −0.5 | 2 | dme-miR-281-2-5p | −0.5 | −1.3 | 0.3 | −0.1 | 0.6 |
| 1 | dme-miR-2b-3p | 1.6 | 1.2 | 0.4 | 0.8 | −1.1 | 2 | dme-miR-2b-2-5p | −0.8 | −1.6 | 0.5 | 1.0 | 0.5 |
| 1 | dme-miR-276a-3p | 3.3 | 0.9 | 1.4 | 1.0 | 0.8 | 2 | dme-miR-375-3p | 0.1 | −2.3 | 0.8 | −0.2 | 0.1 |
| 1 | dme-miR-5-3p | 2.7 | 0.8 | 1.7 | 1.0 | 2.2 | 3 | dme-miR-3-5p | −2.8 | 0.0 | −0.1 | −0.1 | 0.0 |
| 1 | dme-miR-263b-5p | 1.5 | 0.7 | 1.9 | 1.2 | 1.5 | 3 | dme-miR-184-3p | −1.4 | −0.2 | 0.1 | 0.0 | 0.9 |
| 1 | dme-miR-137-3p | 1.3 | −0.6 | 1.6 | 1.3 | 1.4 | 3 | dme-miR-312-5p | −3.8 | −0.2 | −1.1 | −0.6 | −0.6 |
| 1 | dme-miR-1002-5p | 3.1 | −0.2 | 1.2 | 1.9 | 1.4 | 3 | dme-miR-6-1-5p | −4.2 | −0.3 | −0.7 | −0.8 | 0.0 |
| 1 | dme-miR-10-5p | 1.9 | 0.2 | 1.2 | 0.7 | 2.2 | 3 | dme-miR-3-3p | −1.8 | −0.4 | 0.4 | 0.5 | 0.7 |
| 1 | dme-miR-252-5p | 1.5 | 0.8 | 1.7 | 0.7 | 0.5 | 3 | dme-miR-999-3p | −1.7 | −0.4 | −0.2 | −0.1 | 0.6 |
| 1 | dme-miR-316-5p | 3.4 | 0.0 | 1.3 | 0.9 | −0.1 | 3 | dme-miR-305-3p | −1.0 | −0.7 | −0.2 | −0.6 | 0.0 |
| 1 | dme-miR-1006-3p | 2.5 | 0.5 | 1.1 | 0.5 | 0.0 | 3 | dme-miR-9c-5p | −1.7 | −0.8 | −1.0 | −1.0 | 0.0 |
| 1 | dme-miR-968-5p | 4.2 | 0.6 | 1.1 | 0.3 | 0.0 | 3 | dme-miR-13b-2-5p | −1.4 | −0.8 | 0.7 | 0.6 | 0.1 |
| 1 | dme-miR-1000-5p | 1.9 | 0.1 | 1.1 | 0.6 | 0.7 | 3 | dme-miR-9b-3p | −2.5 | −1.1 | 0.0 | −0.1 | −0.3 |
| 1 | dme-miR-987-5p | 2.1 | 0.7 | 0.2 | 0.1 | 1.1 | 3 | dme-miR-79-3p | −2.3 | −1.1 | −0.6 | 0.0 | 0.7 |
| 1 | dme-miR-31a-5p | 1.3 | 0.9 | 0.5 | 0.1 | 0.8 | 3 | dme-miR-92a-3p | −2.3 | −1.3 | 0.3 | −0.3 | 1.4 |
| 1 | dme-miR-13b-3p | 1.1 | 0.1 | 0.9 | 0.7 | 0.5 | 3 | dme-miR-124-5p | −1.5 | −1.3 | 0.5 | 0.5 | 0.4 |
| 1 | dme-miR-927-3p | 1.8 | 0.6 | 0.6 | 0.6 | 0.5 | 3 | dme-miR-1002-3p | −1.6 | −1.6 | 0.3 | 0.2 | 0.0 |
| 1 | dme-miR-996-3p | 1.0 | −0.2 | −0.2 | 0.2 | 0.1 | 3 | dme-miR-308-3p | −1.4 | −1.7 | −0.8 | −0.1 | −0.5 |
| 1 | dme-miR-284-5p | 2.4 | 0.4 | 0.6 | 0.1 | 0.0 | 3 | dme-miR-310-3p | −3.0 | −1.7 | −1.4 | −1.7 | −0.3 |
| 1 | dme-miR-927-5p | 2.1 | −0.6 | 0.5 | 0.4 | 0.0 | 3 | dme-miR-92b-3p | −2.5 | −1.9 | 0.1 | 0.0 | 1.2 |
| 1 | dme-miR-87-3p | 2.6 | −0.3 | 0.7 | 0.0 | −0.1 | 3 | dme-miR-968-3p | −2.1 | −2.2 | 0.3 | 0.5 | 0.7 |
| 1 | dme-miR-281-3p | 1.6 | −0.2 | −0.2 | −0.7 | −0.1 | 3 | dme-miR-995-3p | −1.4 | −2.4 | −0.1 | −0.4 | 0.4 |
| 1 | dme-miR-1008-3p | 1.4 | −0.2 | 0.5 | 0.3 | −0.5 | 3 | dme-miR-312-3p | −5.2 | −2.5 | −1.2 | −0.9 | −0.2 |
| 1 | dme-miR-8-5p | 1.1 | −1.6 | 1.0 | 1.0 | 2.6 | 3 | dme-miR-279-3p | −3.9 | −2.6 | 0.3 | 0.0 | 0.8 |
| 1 | dme-miR-10-3p | 2.2 | −1.0 | 0.6 | 1.2 | 2.0 | 3 | dme-miR-6-2-5p | −4.9 | −2.7 | −1.0 | −0.1 | 0.4 |
| 1 | dme-miR-14-3p | 1.0 | −1.4 | 1.5 | −0.3 | 1.1 | 3 | dme-miR-iab-8-3p | −2.3 | −3.2 | 0.8 | 0.5 | −0.3 |
| 4 | dme-miR-1-3p | −1.5 | 0.2 | 2.2 | 1.2 | 3.8 | 3 | dme-miR-iab-4-5p | −2.3 | −3.3 | 1.0 | 0.7 | −0.3 |
| 4 | dme-miR-988-3p | −1.0 | −0.1 | 1.4 | 1.2 | 0.0 | 3 | dme-miR-275-3p | −2.9 | −3.6 | −1.2 | −1.4 | 0.2 |
| 4 | dme-miR-4-3p | −1.1 | −0.5 | 2.2 | 0.7 | 1.4 | 3 | dme-miR-282-5p | −2.5 | −3.7 | −1.7 | −1.2 | −1.4 |
| 4 | dme-miR-31b-5p | −1.2 | −0.9 | 0.1 | −0.9 | 1.7 | 3 | dme-miR-311-3p | −5.2 | −4.8 | −1.2 | −2.0 | −0.2 |
| 4 | dme-miR-981-3p | 1.0 | −1.9 | −0.1 | 0.6 | 0.4 | 3 | dme-miR-305-5p | −4.0 | −4.9 | −1.8 | −1.8 | −1.3 |
| 2 | dme-miR-9a-5p | 0.8 | 0.5 | 1.9 | 1.5 | 3.3 | 3 | dme-miR-306-5p | −5.1 | −5.2 | −0.6 | −0.8 | −1.1 |
| miRNA | qPCR | RNA-Seq |
|---|---|---|
| dme-miR-1-3p | −1.1 | −1.5 |
| dme-miR-3-3p | −1.5 | −1.8 |
| dme-miR-5-5p | 0.24 | 0.6 |
| dme-miR-7-5p | 1.7 | 2.0 |
| dme-miR-8-3p | 1.3 | 1.1 |
| dme-miR-1002-5p | 3.9 | 3.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosacak, M.I.; Yiğit, H.; Kizil, C.; Akgül, B. Re-Arrangements in the Cytoplasmic Distribution of Small RNAs Following the Maternal-to-Zygotic Transition in Drosophila Embryos. Genes 2018, 9, 82. https://doi.org/10.3390/genes9020082
Cosacak MI, Yiğit H, Kizil C, Akgül B. Re-Arrangements in the Cytoplasmic Distribution of Small RNAs Following the Maternal-to-Zygotic Transition in Drosophila Embryos. Genes. 2018; 9(2):82. https://doi.org/10.3390/genes9020082
Chicago/Turabian StyleCosacak, Mehmet Ilyas, Hatice Yiğit, Caghan Kizil, and Bünyamin Akgül. 2018. "Re-Arrangements in the Cytoplasmic Distribution of Small RNAs Following the Maternal-to-Zygotic Transition in Drosophila Embryos" Genes 9, no. 2: 82. https://doi.org/10.3390/genes9020082
APA StyleCosacak, M. I., Yiğit, H., Kizil, C., & Akgül, B. (2018). Re-Arrangements in the Cytoplasmic Distribution of Small RNAs Following the Maternal-to-Zygotic Transition in Drosophila Embryos. Genes, 9(2), 82. https://doi.org/10.3390/genes9020082