Analysis of Gene Regulatory Networks of Maize in Response to Nitrogen


Abstract
:1. Introduction
2. Material and Methods
2.1. Microarray Data
2.2. Differential Expression Analysis and Comparison
2.3. The Steps for Identifying Potential Regulators
2.4. Mapping Differentially Expressed Genes onto Known Networks
2.5. Artificial Neural Network Inference
2.6. Mutual Information Network Inference
2.7. Plant Materials
2.8. RNA Extraction
2.9. qRT-PCR Analysis
3. Results
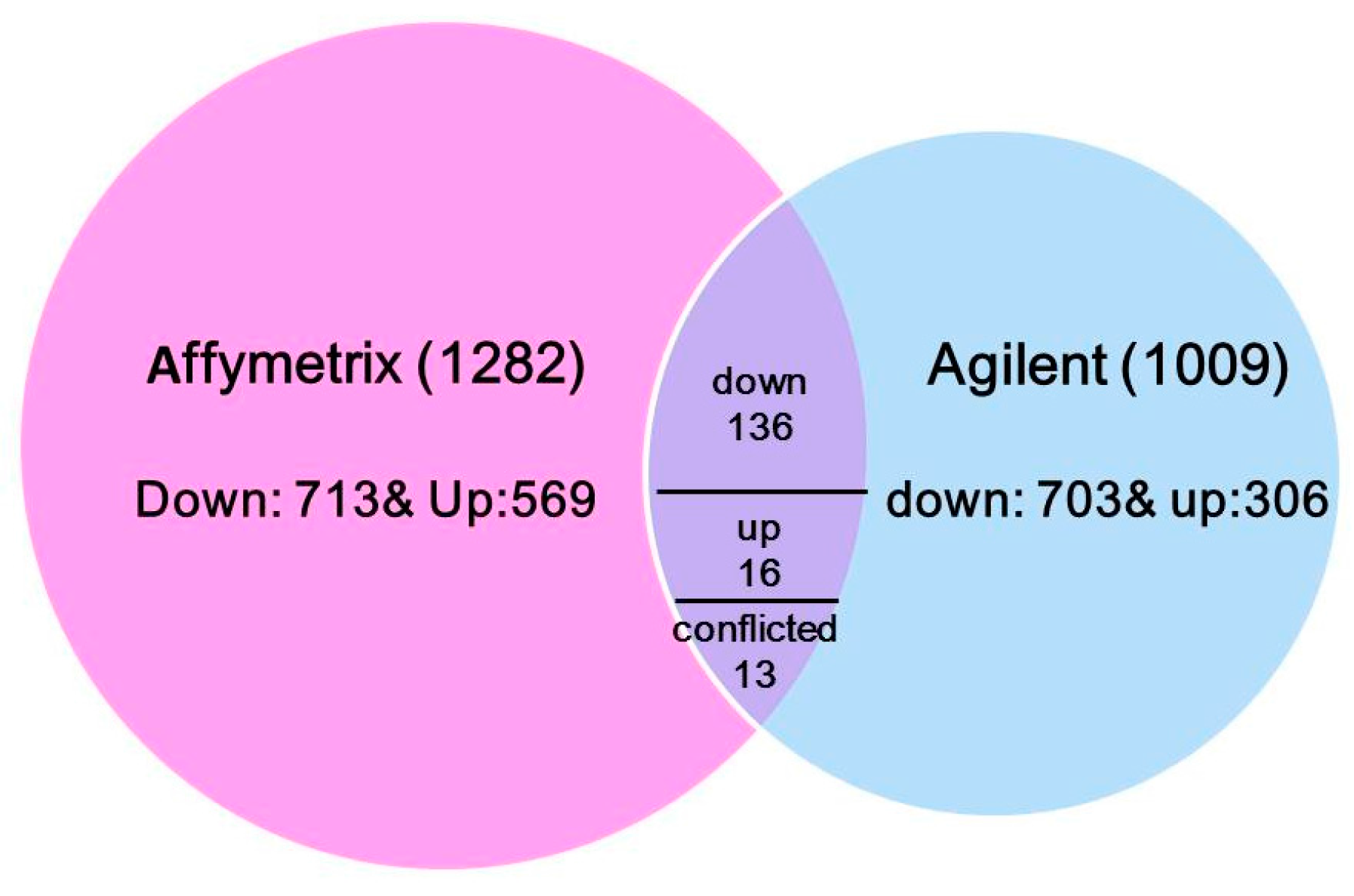
3.1. Comparison of Differentially Expressed N-Responsive Genes across Two Microarray Datasets
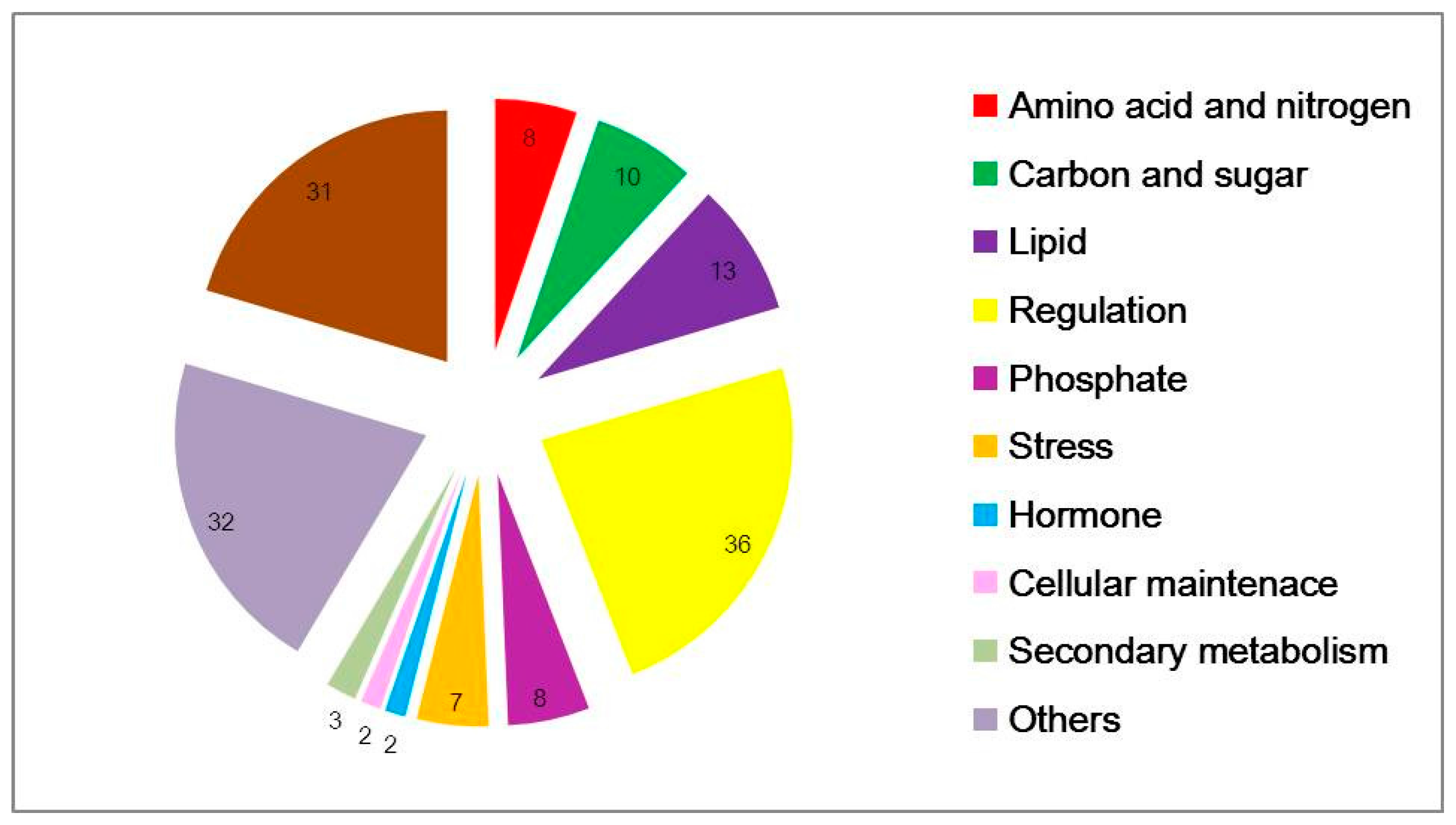
3.2. Subnetworks from Known Networks
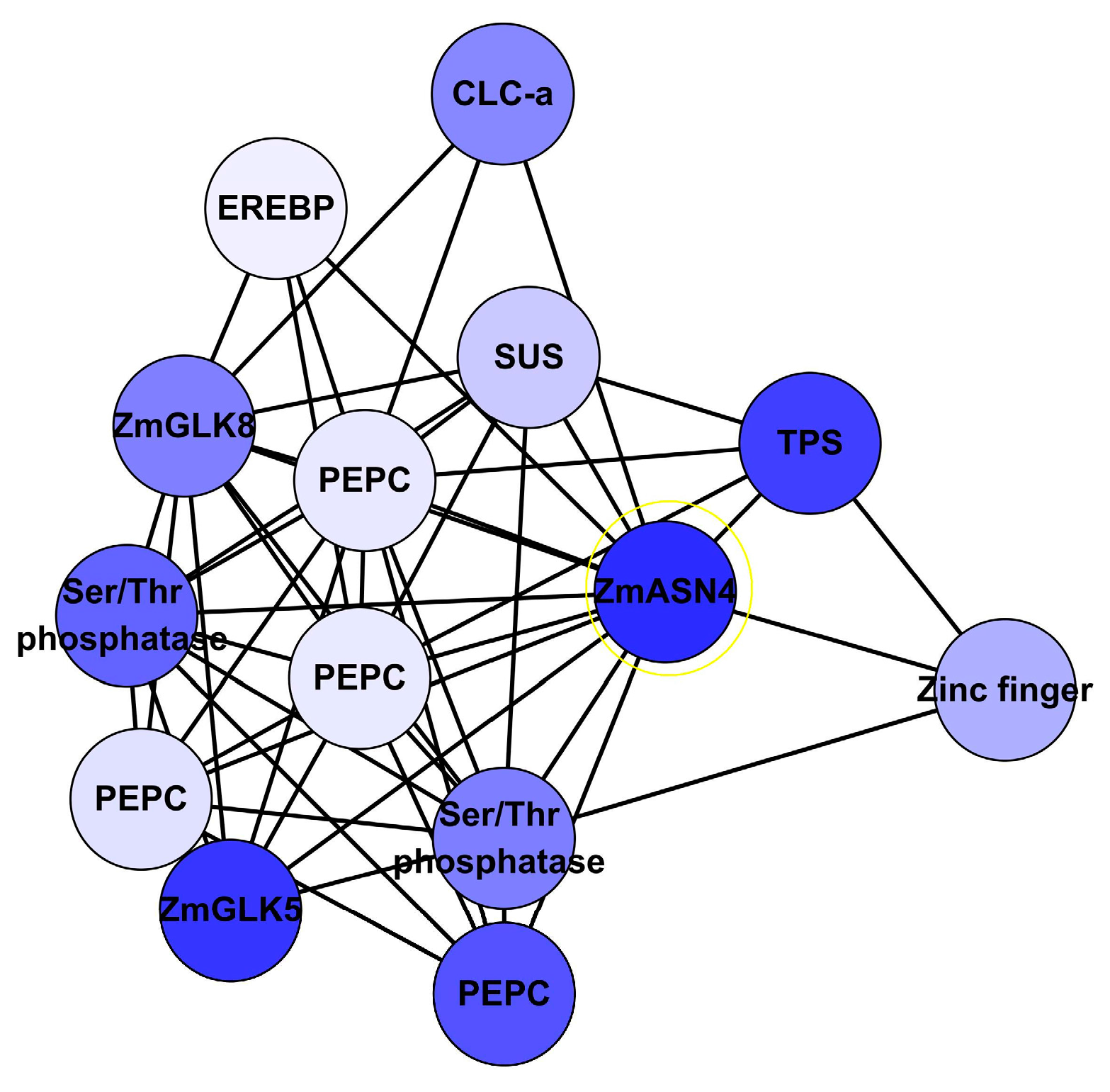
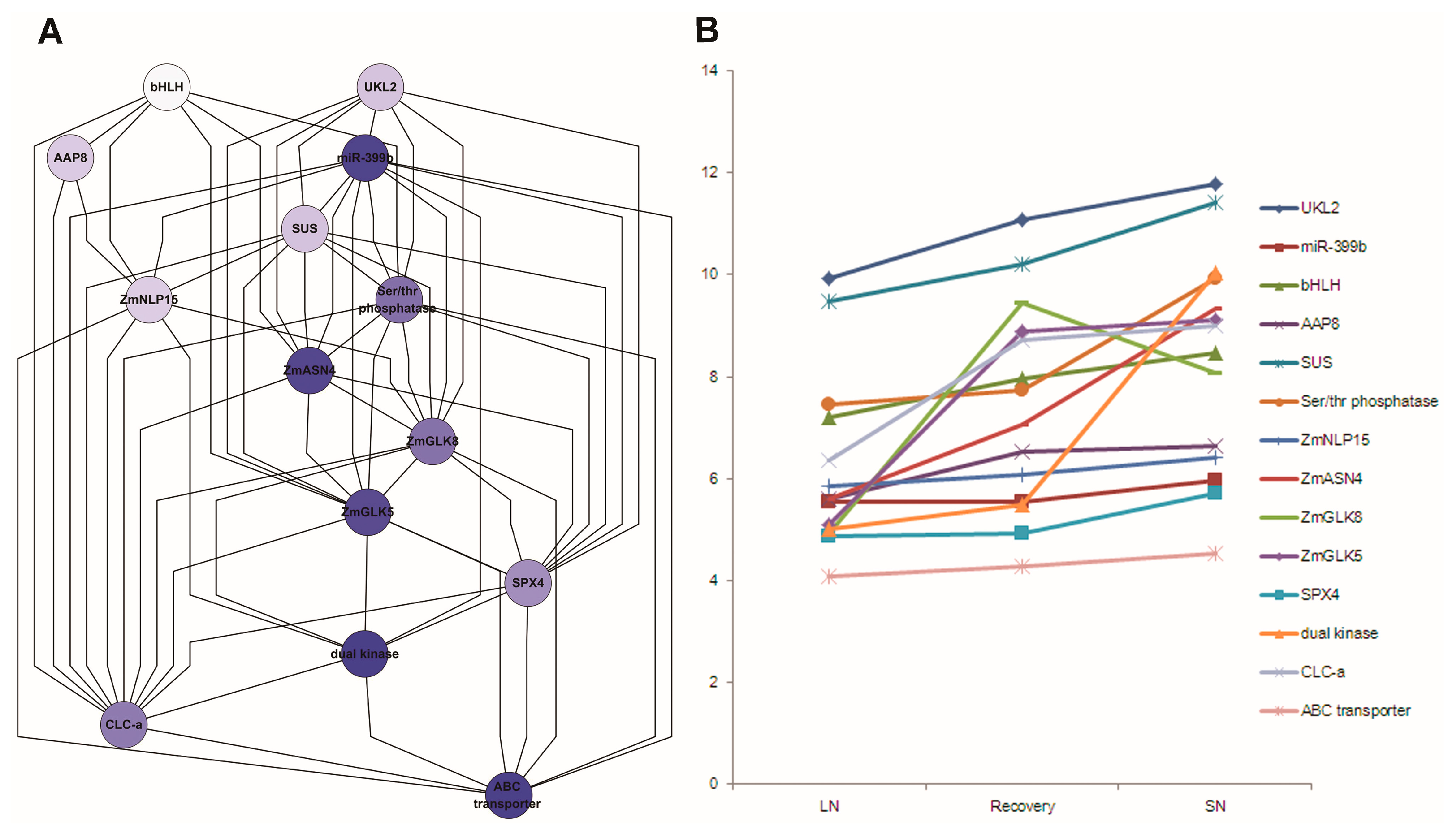
3.3. Identification of N-Responsive Genes Using Artificial Neural Network Analysis
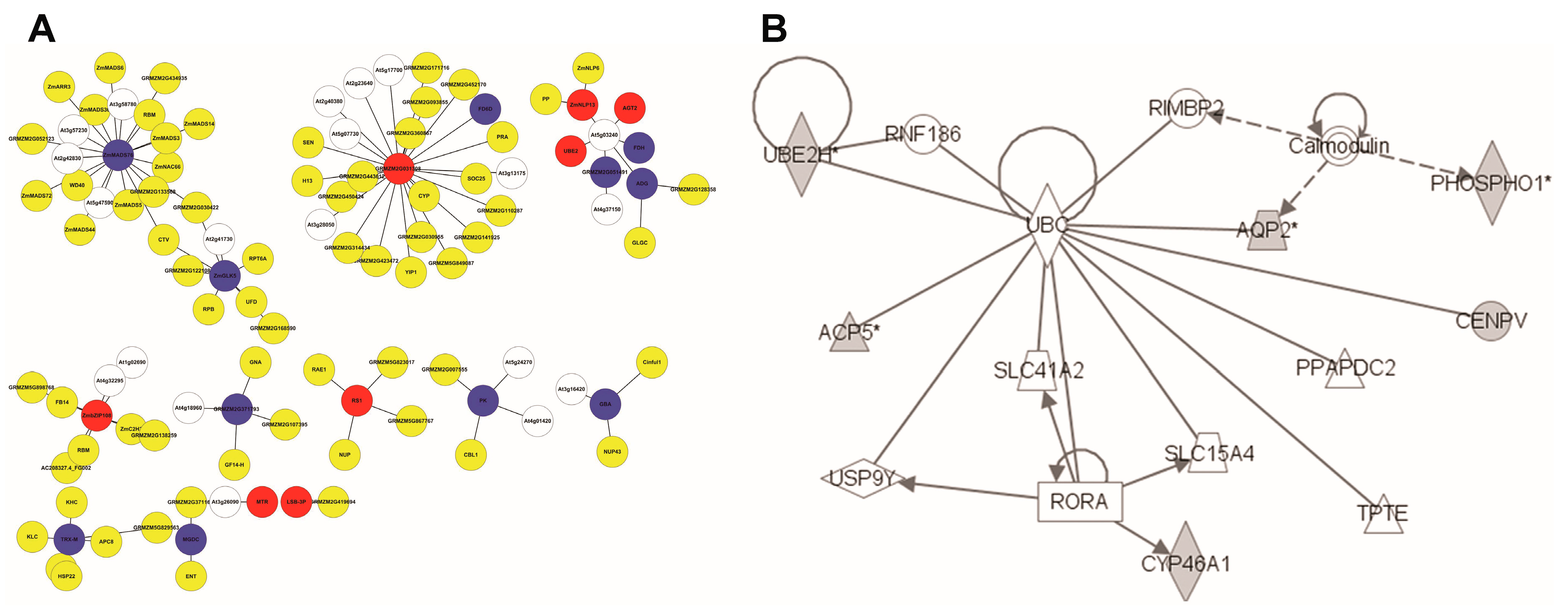
3.4. Identification of N-Responsive Transcription Factors Using Artificial Neural Network Analysis
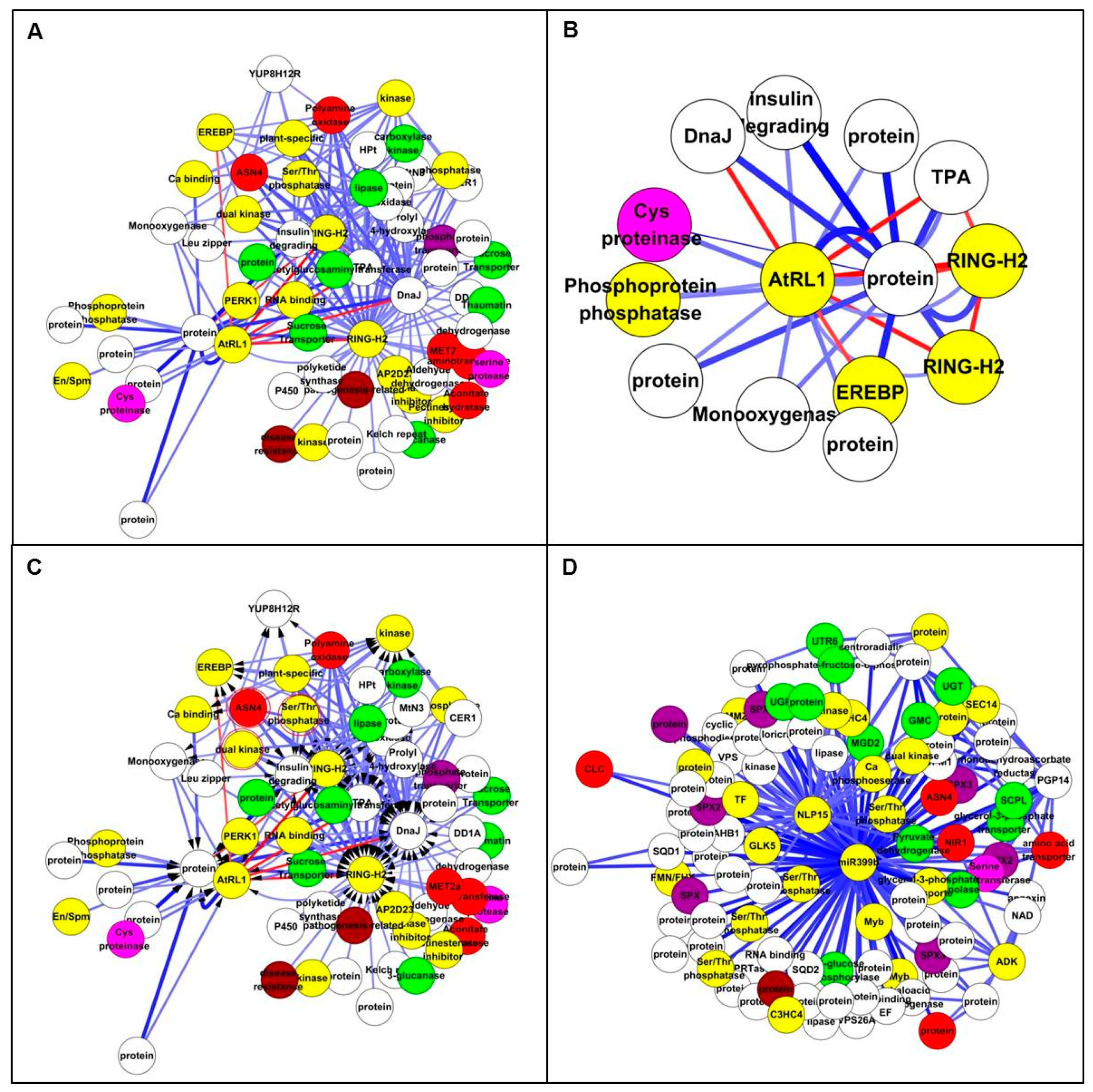
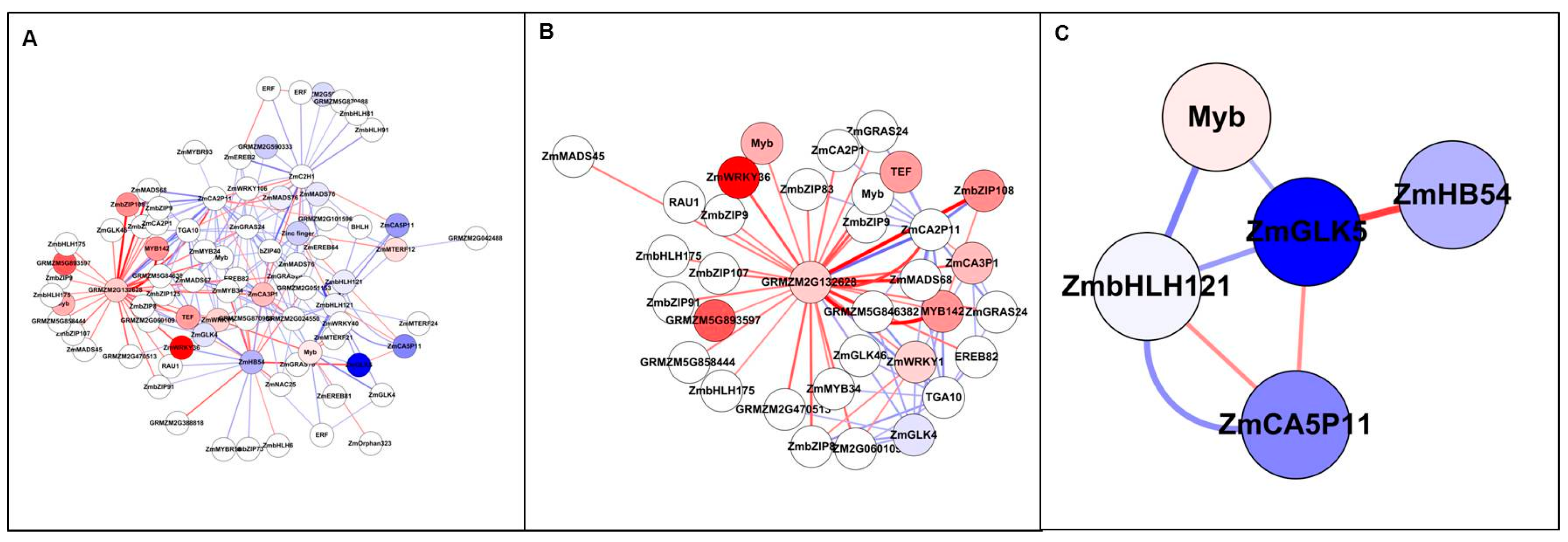
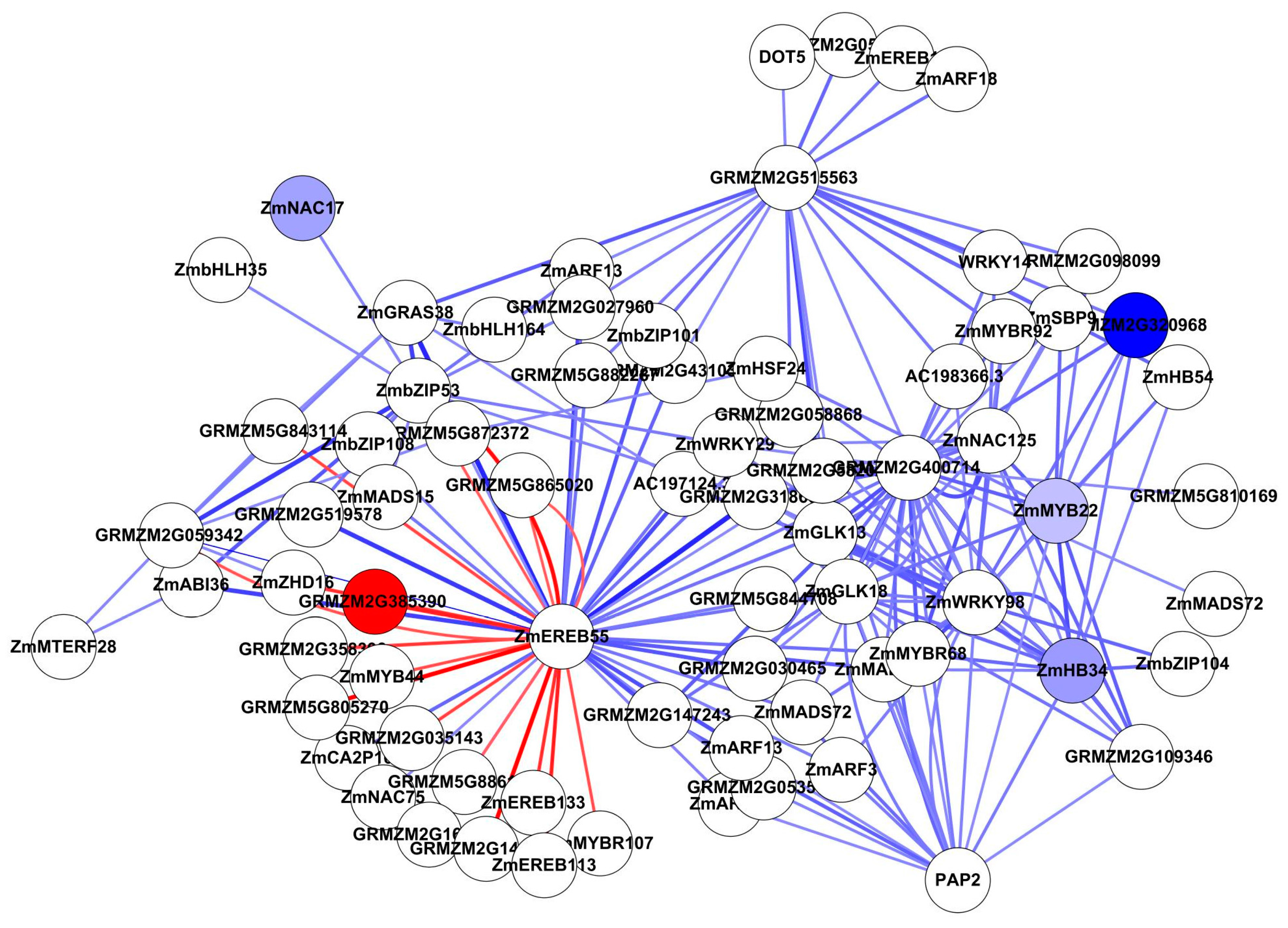
3.5. Analysis of Genome-Scale Networks—Subnetworks from All Genes Input
3.6. Analysis of Genome-Scale Networks—Subnetworks from Differentially Expressed Gene Input
3.7. qRT-PCR Analysis for Selected Candidate Genes
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goulding, K. Nitrate leaching from arable and horticultural land. Soil Use Manag. 2000, 16, 145–151. [Google Scholar] [CrossRef]
- Cassman, K.G.; Dobermann, A.; Walters, D.T. Agroecosystems, nitrogen-use efficiency, and nitrogen management. AMBIO 2002, 31, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Agrama, H.A.S.; Zakaria, A.G.; Said, F.B.; Tuinstra, M. Identification of quantitative trait loci for nitrogen use efficiency in maize. Mol. Breed. 1999, 5, 187–195. [Google Scholar] [CrossRef]
- Bertin, P.; Gallais, A. Genetic variation for nitrogen use efficiency in a set of recombinant maize inbred lines. I. Agrophysiological results. Maydica 2000, 45, 53–66. [Google Scholar]
- Hirel, B.; Bertin, P.; Quillere, I.; Bourdoncle, W.; Attagnant, C.; Dellay, C.; Gouy, A.; Cadiou, S.; Retailliau, C.; Falque, M.; et al. Towards a better understanding of the genetic and physiological basis for nitrogen use efficiency in maize. Plant Physiol. 2001, 125, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Guegler, K.; LaBrie, S.T.; Crawford, N.M. Genomic analysis of a nutrient response in Arabidopsis reveals diverse expression patterns and novel metabolic and potential regulatory genes induced by nitrate. Plant Cell 2000, 12, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Okamoto, M.; Xing, X.J.; Crawford, N.M. Microarray analysis of the nitrate response in Arabidopsis roots and shoots reveals over 1000 rapidly responding genes and new linkages to glucose, trehalose-6-phosphate, iron, and sulfate metabolism. Plant Physiol. 2003, 132, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, R.A.; Lejay, L.V.; Dean, A.; Chiaromonte, F.; Shasha, D.E.; Coruzzi, G.M. Qualitative network models and genome-wide expression data define carbon/nitrogen-responsive molecular machines in Arabidopsis. Genome Biol. 2007, 8, R7. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.M.; Kant, S.; Clark, J.; Gidda, S.; Ming, F.; Xu, J.Y.; Rochon, A.; Shelp, B.J.; Hao, L.X.; Zhao, R.; et al. Increased nitrogen-use efficiency in transgenic rice plants over-expressing a nitrogen-responsive early nodulin gene identified from rice expression profiling. Plant Cell Environ. 2009, 32, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Y.; Qian, Q.; Wu, K.; Luo, J.L.; Wang, S.S.; Zhang, C.W.; Ma, Y.F.; Liu, Q.; Huang, X.Z.; Yuan, Q.B.; et al. Heterotrimeric G proteins regulate nitrogen-use efficiency in rice. Nat. Genet. 2014, 46, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Konishi, M.; Yanagisawa, S. Arabidopsis NIN-like transcription factors have a central role in nitrate signalling. Nat. Commun. 2013, 4, 1617. [Google Scholar] [CrossRef] [PubMed]
- Kurai, T.; Wakayama, M.; Abiko, T.; Yanagisawa, S.; Aoki, N.; Ohsugi, R. Introduction of the ZmDof1 gene into rice enhances carbon and nitrogen assimilation under low-nitrogen conditions. Plant Biotechnol. J. 2011, 9, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Marchive, C.; Roudier, F.; Castaings, L.; Bréhaut, V.; Blondet, E.; Colot, V.; Meyer, C.; Krapp, A. Nuclear retention of the transcription factor NLP7 orchestrates the early response to nitrate in plants. Nat. Commun. 2013, 4, 1713. [Google Scholar] [CrossRef] [PubMed]
- Rubio, V.; Linhares, F.; Solano, R.; Martín, A.C.; Iglesias, J.; Leyva, A.; Paz-Ares, J. A conserved MYB transcription factor involved in phosphate starvation signaling both in vascular plants and in unicellular algae. Genes Dev. 2001, 15, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Guzman, P. ATLs and BTLs, plant-specific and general eukaryotic structurally-related E3 ubiquitin ligases. Plant Sci. 2014, 215–216, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Wagner, A.; Kos, A.; Aschrafi, A.; Handrick, R.; Hannemann, J.; Otte, K. Breaking limitations of complex culture media: Functional non-viral miRNA delivery into pharmaceutical production cell lines. J. Biotechnol. 2013, 168, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.A.; Araus, V.; Lu, C.; Parry, G.; Green, P.J.; Coruzzi, G.M.; Gutiérrez, R.A. Nitrate-responsive miR393/AFB3 regulatory module controls root system architecture in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 4477–4482. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, S.; Begheldo, M.; Nonis, A.; Quaggiotti, S. The miRNA-mediated post-transcriptional regulation of maize response to nitrate. Plant Signal. Behav. 2012, 7, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, R.S.; Lin, H.; Childs, K.L.; Hansey, C.N.; Buell, C.R.; Leon, N.D.; Kaeppler, S.M. Genome-wide atlas of transcription during maize development. Plant J. 2011, 66, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Amiour, N.; Imbaud, S.; Clément, G.; Agier, N.; Zivy, M.; Valot, B.; Balliau, T.; Armengaud, P.; Quilleré, I.; Canas, R.; et al. The use of metabolomics integrated with transcriptomic and proteomic studies for identifying key steps involved in the control of nitrogen metabolism in crops such as maize. J. Exp. Bot. 2012, 63, 5017–5033. [Google Scholar] [CrossRef] [PubMed]
- Downs, G.S.; Bi, Y.M.; Colasanti, J.; Wu, W.Q.; Chen, X.; Zhu, T.; Rothstein, S.J.; Lukens, L.N. A developmental transcriptional network for maize defines coexpression modules. Plant Physiol. 2013, 161, 1830–1843. [Google Scholar] [CrossRef] [PubMed]
- Humbert, S.; Subedi, S.; Cohn, J.; Zeng, B.; Bi, Y.M.; Chen, X.; Zhu, T.; McNicholas, P.D.; Rothstein, S.J. Genome-wide expression profiling of maize in response to individual and combined water and nitrogen stresses. BMC Genom. 2013, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, A.; Astolfi, S.; Zuchi, S.; Pii, Y.; Guardini, K.; Tononi, P.; Varanini, Z. Nitrate induction triggers different transcriptional changes in a high and a low nitrogen use efficiency maize inbred line. J. Integr. Plant Biol. 2014, 56, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.S.; Wu, J.R.; Ziegler, T.E.; Yang, X.; Zayed, A.; Rajani, M.S.; Zhou, D.F.; Basra, A.S.; Schachtman, D.P.; Peng, M.S.; et al. Gene expression biomarkers provide sensitive indicators of in planta nitrogen status in maize. Plant Physiol. 2010, 157, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, U.; Mascher, M.; Colmsee, C.; Scholz, U.; Bräutigam, A.; Fahnenstich, H.; Sonnewald, U. Maize source leaf adaptation to nitrogen deficiency affects not only nitrogen and carbon metabolism but also control of phosphate homeostasis. Plant Physiol. 2012, 160, 1384–1406. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Kellis, M.; Collins, J.J.; Stolovitzky, G. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bradley, G.; Pyke, K.; Ball, G.; Lu, C.G.; Fray, R.; Marshall, A.; Jayasuta, S.; Baxter, C.; Wijk, R.V.; et al. Network inference analysis identifies an APRR2-like gene linked to pigment accumulation in tomato and pepper fruits. Plant Physiol. 2013, 161, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; Kersey, P.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Bolser, D.; Staines, D.M.; Pritchard, E.; Kersey, P. Ensembl Plants: Integrating tools for visualizing, mining, and analyzing plant genomics data. Methods Mol Biol. 2016, 1374, 115–140. [Google Scholar] [PubMed]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Lancashire, L.J.; Lemetre, C.; Ball, G.R. An introduction to artificial neural networks in bioinformatics—Application to complex microarray and mass spectrometry datasets in cancer studies. Brief. Bioinform. 2009, 10, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wei, H.R.; Zhao, P.X. DeGNServer: Deciphering genome-scale gene networks through high performance reverse engineering analysis. BioMed Res. Int. 2013, 2013, 856325. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, X.Y.; Chen, W.J.; Peng, X.J.; Chen, X.; Zhu, S.W.; Cheng, B.J. Whole-genome survey and characterization of MADS-box gene family in maize and sorghum. Plant Cell Tissue Org. Cult. 2011, 105, 159–173. [Google Scholar] [CrossRef]
- Mascheretti, I.; Battaglia, R.; Mainier, D.; Altana, A.; Lauria, M.; Rossi, V. The WD40-repeat proteins NFC101 and NFC102 regulate different aspects of maize development through chromatin modification. Plant Cell 2014, 25, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Rossini, L.; Cribb, L.; Martin, D.J.; Langdale, J.A. The maize Golden2 gene defines a novel class of transcriptional regulators in plants. Plant Cell 2001, 13, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Obertello, M.; Krouk, G.; Katari, M.S.; Runko, S.J.; Coruzzi, G.M. Modeling the global effect of the basic-leucine zipper transcription factor 1 (bZIP 1) on nitrogen and light regulation in Arabidopsis. BMC Syst. Biol. 2011, 4, 111. [Google Scholar]
- Criqui, M.C.; Engler, J.D.A.; Camasses, A.; Capron, A.; Paemerntier, Y.; Inzé, D.; Genschik, P. Molecular characterization of plant ubiquitin-conjugating enzymes belonging to the UbcP4/E2-C/UBCx/UbcH10 gene family. Plant Physiol. 2002, 130, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Vourc’h, P.; Mahé, M.; Thépault, A.; Antar, C.; Védrine, S.; Praline, J.; Camu, W.; Andres, C.R.; Corcia, P.; et al. Association study of the ubiquitin conjugating enzyme gene UBE2H in sporadic ALS. Amyotroph. Lateral Scler. 2009, 10, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, Y.; Meaney, S.; Heverin, M.; Ekström, L.; Brafaman, A.; Shafir, M.; Andersson, U.; Olin, M.; Eggertsen, G.; Diczfalusy, U.; et al. Studies on the transcriptional regulation of cholesterol 24-hydroxylase (CYP46A1): Marked insensitivity toward different regulatory axes. J. Biol. Chem. 2006, 281, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, Y.; Li, Y.; Zheng, J.; Tang, J. A novel RING finger E3 ligase RNF186 regulate ER stress-mediated apoptosis through interaction with BNip1. Cell. Signal. 2013, 25, 2320–2333. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; McAvoy, S.; Kuhn, R.; Smith, D.I. RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene 2006, 25, 2901–2908. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.I.; Chiang, S.F.; Lin, W.Y.; Chen, J.W.; Tseng, C.Y.; Wu, P.C.; Chiou, T.J. Regulatory network of microRNA399 and PHO2 by systemic signaling. Plant Physiol. 2008, 147, 732–746. [Google Scholar] [CrossRef] [PubMed]
- De Angeli, A.; Monachello, D.; Ephritikhine, G.; Frachissem, J.M.; Thomine, S.; Gambale, F.; Barbier-Brygoo, H. The nitrate/proton antiporter AtCLCa mediates nitrate accumulation in plant vacuoles. Nature 2006, 442, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Morot-Gaudry-Talarmain, Y.; Rockel, P.; Moureaux, T.; Quillere, I.; Leydecker, M.T.; Kaiser, W.M.; Morot-Gaudry, J.F. Nitrite accumulation and nitric oxide emission in relation to cellular signaling in nitrite reductase antisense tobacco. Planta 2002, 215, 708–715. [Google Scholar] [PubMed]
- Rushton, D.L.; Tripathi, P.; Rabara, R.C.; Lin, J.; Ringler, P.; Boken, A.K.; Langum, T.J.; Smidt, L.; Boomsma, D.D.; Emme, N.J.; et al. WRKY transcription factors: key components in abscisic acid signalling. Plant Biotechnol. J. 2012, 10, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Cañas, R.A.; Quilleré, I.; Lea, P.J.; Hirel, B. Analysis of amino acid metabolism in the ear of maize mutants deficient in two cytosolic glutamine synthetase isoenzymes highlights the importance of asparagine for nitrogen translocation within sink organs. Plant Biotechnol. J. 2010, 8, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.; Screen, S.; Crowley, J.; Peng, J.X.; Andersen, S.; Brown, T.; Qi, Q.G.; Fabbri, B.; Duff, S.M. Identification and characterization of four distinct asparagine synthetase (AsnS) genes in maize (Zea mays L.). Plant Sci. 2008, 175, 799–808. [Google Scholar] [CrossRef]
- Gonzalez, D.H.; Iglesias, A.A.; Andreo, C.S. Active-site-directed inhibition of phosphoenolpyruvate carboxylase from maize leaves by bromopyruvate. Arch. Biochem. Biophys. 1986, 245, 179–186. [Google Scholar] [CrossRef]
- Nimmo, H.G. The regulation of phosphoenolpyruvate carboxylase in CAM plants. Trends Plant Sci. 2000, 5, 75–80. [Google Scholar] [CrossRef]
- Quick, P.; Siegl, G.; Neuhaus, E.; Feil, R.; Stitt, M. Short-term water stress leads to a stimulation of sucrose synthesis by activating sucrose-phosphate synthase. Planta 1989, 177, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.L.; Huber, J.L.A.; Huber, S.C. Sucrose phosphate synthase and sucrose accumulation at low temperature. Plant Physiol. 1992, 100, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.J.; Minchin, F.R.; Skot, L.; James, C.L. Stress-induced declines in soybean N2 fixation are related to nodule sucrose synthase activity. Plant Physiol. 1997, 114, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Benning, C.; Ohta, H. Three enzyme systems for galactoglycerolipid biosynthesis are coordinately regulated in plants. J. Biol. Chem. 2005, 280, 2397–2400. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Wong, P.; Chan, H.K.; Yam, K.M.; Chen, L.; Chow, C.M.; Coruzzi, G.M. Overexpression of the ASN1 gene enhances nitrogen status in seeds of Arabidopsis. Plant Physiol. 2003, 132, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Water, M.T.; Moylan, E.C.; Langdale, J.A. GLK transcription factors regulate chloroplast development on a cell-autonomous manner. Plant J. 2008, 56, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Schauser, L.; Roussis, A.; Stiller, J.; Stougaard, J. A plant regulator controlling development of symbiotic root nodules. Nature 1999, 402, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhu, J.K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed]
- Bari, R.; Pant, B.D.; Stitt, M.; Scheible, W.R. PHO2, microRNA399, and PHR1 define a phosphate-signaling pathway in plants. Plant Physiol. 2006, 141, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, M.; Shi, B.J.; Gustafson, P.; Langridge, P. Characterization of phosphorus-regulated miR399 and miR827 and their isomirs in barley under phosphorus-sufficient and phosphorus-deficient conditions. BMC Plant Biol. 2013, 13, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study 1 | Study 2 | |
|---|---|---|
| Authors | Yang et al., 2011 [24] | Schluter et al., 2012 [25] |
| Aim | Identify biomarkers of Nitrogen status | Leaf responses to low Nitrogen |
| Platform | Affymetrix array | Agilent array |
| Plant varieties | 4 commercial hybrids | Inbred A188 and B73 |
| Growth conditions | Greenhouse | Growth chamber (14 h light + 10 h dark) |
| Sufficient N | 20 mM NH4NO3 | 15 mM KNO3 |
| Low N | 2 mM NH4NO3 | 0.15 mM KNO3 |
| Harvest | Plants sown so that all sample were harvested on same day. V6 leaves taken (at 21 days for sufficient N and 28 days for low N) plus following day for Line 4 and N-recovered plants. 3 replicates. 4 plants per replicate. Material harvested at 10 am and 11 pm on both days. | Material from 2 identical experiments combined. V5 and V6 leaves taken respectively at 20 and 30 days. 4 replicates. 1 plant from each experiment per replicate. Material harvested after 2 h of light. |
| Microarray | Total Probes | Mapped Probes (Genes) | DE Probes (Genes) FC > 1.5 | Down Regulated Genes | Up Regulated Genes | Conflicted Genes |
|---|---|---|---|---|---|---|
| Study 1 | 84,246 | 52,304 (31,958) | 1282 (1088) | |||
| Study 2 | 41,838 | 35,086 (27,278) | 1009 (923) | |||
| Common genes | □ | (19,991) | 165 | 136 | 16 | 13 |
| Microarray | Genotype | Differentially Regulated Transcripts | |||||
|---|---|---|---|---|---|---|---|
| Down | Up | Total | Retrieved Down | Retrieved Up | Conflicted | ||
| Study 1 | Line 1 | 1268 | 2391 | 3659 | |||
| Line 2 | 1194 | 827 | 2021 | ||||
| Line 3 | 914 | 1529 | 2443 | ||||
| Line 4 | 1329 | 1093 | 2422 | ||||
| Lines 1–4 | 713 | 569 | 1282 | 596 | 492 | ||
| Study 2 | A188 (D30) | 1506 | 675 | 2181 | |||
| B73 (D30) | 1608 | 1474 | 3082 | ||||
| A188 and B73 (D30) | 703 | 306 | 1009 | 639 | 284 | ||
| Lines 1 and 3, and A188 | 184 | 39 | 30 | ||||
| Lines 1 and 3, and B73 | 158 | 66 | 70 | ||||
| Lines 2 and 4, and A188 | 240 | 25 | 22 | ||||
| Lines 2 and 4, and B73 | 203 | 40 | 56 | ||||
| Studies 1 and 2 | Lines 1–4, A188, and B73 | 136 | 16 | 13 | |||
| MaizeGDB Gene | Affy ID | Agi ID | FC | Gene Description |
|---|---|---|---|---|
| GRMZM2G141320 | A1ZM005708_at | P_OptiV1C15662 | −97.7 | UDP-galactosyltransferase |
| GRMZM2G333224 | A1ZM067520_at | P_OptiV1S29808 | −64.4 | abc transporter |
| GRMZM2G018820 | A1ZM001292_s_at | P_OptiV1C11631 | −45.6 | GDE1 |
| GRMZM2G016370 | A1ZM060059_at | P_OptiV1C16142 | −39.1 | ZmGLK5 |
| GRMZM2G100454 | A1ZM062303_at | P_OptiV1C09974 | −35.3 | dual-specific kinase |
| GRMZM2G373607 | A1ZM045918_at | P_OptiV1S32106 | −32.2 | SCPL |
| GRMZM2G086179 | A1ZM005813_s_at | P_OptiV1N40557 | −28.6 | |
| zma-MIR399b | A1ZM015359_at | P_OptiV1N41856 | −26.4 | zma-MIR399b |
| GRMZM2G176562 | A1ZM019400_at | P_OptiV1S17969 | −24.9 | SQD2 |
| GRMZM2G060311 | A1ZM015149_at | P_OptiV1S18616 | −24.6 | hydrophobic protein OSR8-like |
| GRMZM2G152447 | A1ZM002117_at | P_OptiV1C10994 | −22.0 | Purple acid phosphatase 1 |
| GRMZM2G526727 | A1ZM055955_at | P_OptiV1C13366 | −21.0 | Arginine decarboxylase |
| GRMZM5G805389 | A1ZM017973_at | P_OptiV1C03719 | −19.6 | SPX domain |
| GRMZM5G853702 | A1ZM038244_at | P_OptiV1N38765 | −19.2 | bowman-birk type trypsin inhibitor precursor |
| GRMZM2G043565 | A1ZM013564_at | P_OptiV1N41328 | −18.6 | |
| GRMZM2G035579 | A1ZM024880_at | P_OptiV1N39672 | −16.6 | spx domain-containing protein |
| GRMZM2G078633 | A1ZM058393_at | P_OptiV1C00703 | −16.4 | asparagine synthase (ZmASN4) |
| GRMZM2G344654 | A1ZM003814_s_at | P_OptiV1C00507 | −16.1 | |
| GRMZM2G049541 | A1ZM055541_at | P_OptiV1C04552 | −16 | phosphoenolpyruvate carboxylase kinase |
| GRMZM2G069694 | A1ZM006239_at | P_OptiV1C03616 | −15.8 | Plant specific |
| GRMZM5G816348 | A1ZM062346_at | P_OptiV1C03692 | 10.3 | oligopeptide transporter |
| GRMZM2G355752 | A1ZM078046_x_at | P_OptiV1S20690 | 7.6 | early light-induced protein |
| GRMZM2G087507 | A1ZM056359_at | P_OptiV1C01116 | 6.3 | probable nad h-dependent oxidoreductase |
| GRMZM2G177098 | A1ZM000024_a_at | P_OptiV1C14892 | 5.1 | terpene synthase |
| GRMZM2G330635 | A1ZM011141_at | P_OptiV1S29121 | 4.7 | glutathione s-transferase |
| GRMZM2G455582 | A1ZM069754_at | P_OptiV1S29347 | 4.2 | pentatricopeptide repeat |
| AC185430.3 | A1ZM023429_at | P_OptiV1N39003 | 3.7 | Rho guanyl-nucleotide |
| GRMZM5G812407 | A1ZM055479_at | P_OptiV1C10233 | 3.6 | ZmC3H51 |
| GRMZM5G857944 | A1ZM003474_a_at | P_OptiV1C16320 | 3.5 | ZmCA2P13 |
| GRMZM2G367907 | A1ZM003724_s_at | P_OptiV1N39816 | 3.3 | IMP dehydrogenase |
| GRMZM2G032807 | A1ZM055038_s_at | P_OptiV1S28735 | 3.1 | heavy metal-associated domain |
| GRMZM2G359664 | A1ZM027751_at | P_OptiV1S21353 | 3.1 | pollen-specific kinase |
| GRMZM2G016323 | A1ZM046170_s_at | P_OptiV1N38397 | 3.0 | ubiquitin carboxyl-terminal hydrolase |
| GRMZM2G177169 | A1ZM005058_at | P_OptiV1S24357 | 2.4 | pentatricopeptide repeat |
| GRMZM2G440349 | A1ZM041153_at | P_OptiV1S27652 | 2.4 | pentatricopeptide repeat |
| GRMZM2G462625 | A1ZM012815_at | P_OptiV1S23216 | 2.3 | pentatricopeptide repeat |
| MaizeGDB Gene ID | Affy ID | Agi ID | log2FC (Affy) | log2FC (Agi) | Gene Description |
|---|---|---|---|---|---|
| GRMZM2G100454 | A1ZM062303_at | P_OptiV1C09974 | −64.9 | −18.9 | Putative dual-specific kinase |
| GRMZM2G134054 | A1ZM057368_s_at | P_OptiV1S25751 | −11.6 | −10.6 | Ser/Thr protein phosphatase |
| GRMZM2G078633 | A1ZM058393_at | P_OptiV1C00703 | −34.1 | −7.9 | ZmASN4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Ball, G.; Hodgman, C.; Coules, A.; Zhao, H.; Lu, C. Analysis of Gene Regulatory Networks of Maize in Response to Nitrogen. Genes 2018, 9, 151. https://doi.org/10.3390/genes9030151
Jiang L, Ball G, Hodgman C, Coules A, Zhao H, Lu C. Analysis of Gene Regulatory Networks of Maize in Response to Nitrogen. Genes. 2018; 9(3):151. https://doi.org/10.3390/genes9030151
Chicago/Turabian StyleJiang, Lu, Graham Ball, Charlie Hodgman, Anne Coules, Han Zhao, and Chungui Lu. 2018. "Analysis of Gene Regulatory Networks of Maize in Response to Nitrogen" Genes 9, no. 3: 151. https://doi.org/10.3390/genes9030151
APA StyleJiang, L., Ball, G., Hodgman, C., Coules, A., Zhao, H., & Lu, C. (2018). Analysis of Gene Regulatory Networks of Maize in Response to Nitrogen. Genes, 9(3), 151. https://doi.org/10.3390/genes9030151