Identification of a WNT5A-Responsive Degradation Domain in the Kinesin Superfamily Protein KIF26B

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Protein Mutagenesis
2.3. Generation of Stable NIH/3T3 Lines
2.4. Molecular Cloning of KIF26A-C
2.5. Recombinant Proteins and Inhibitors
2.6. WNT5A Stimulation and Flow Cytometry
2.7. Lentivirus-Mediated Protein Expression
2.8. Protein Sequence Analysis
3. Results
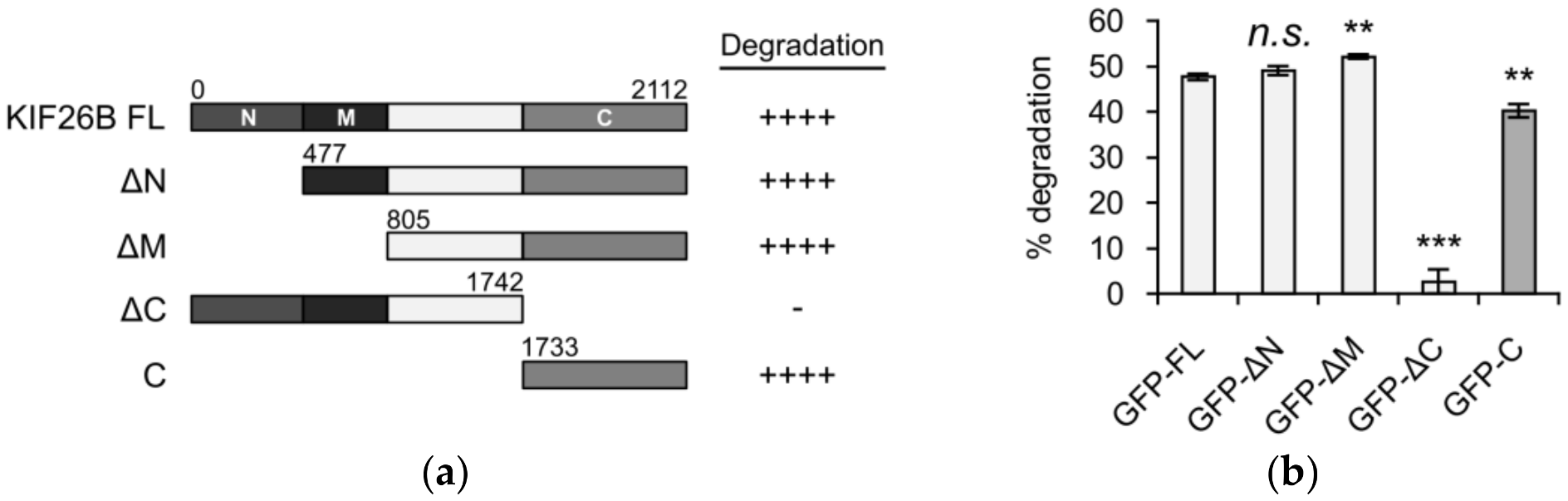
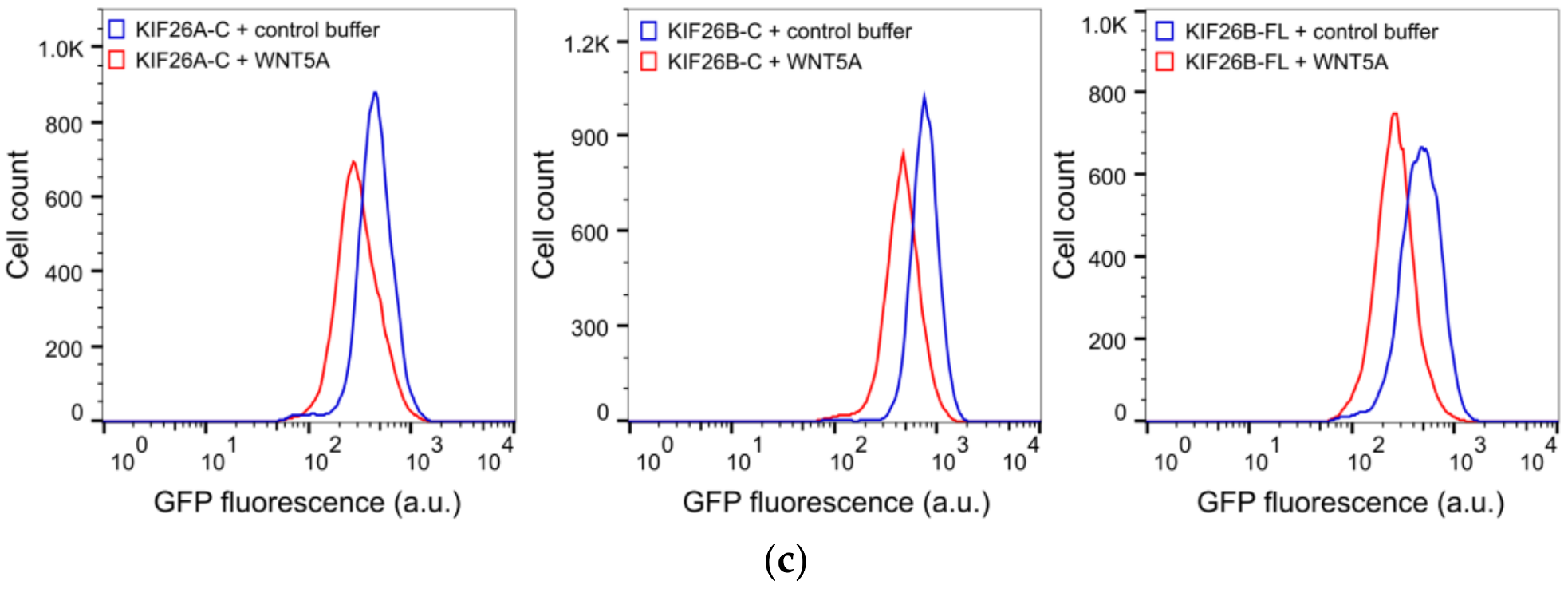
3.1. The C-Terminus of KIF26B Contains a WNT5A-Responsive Degradation Domain
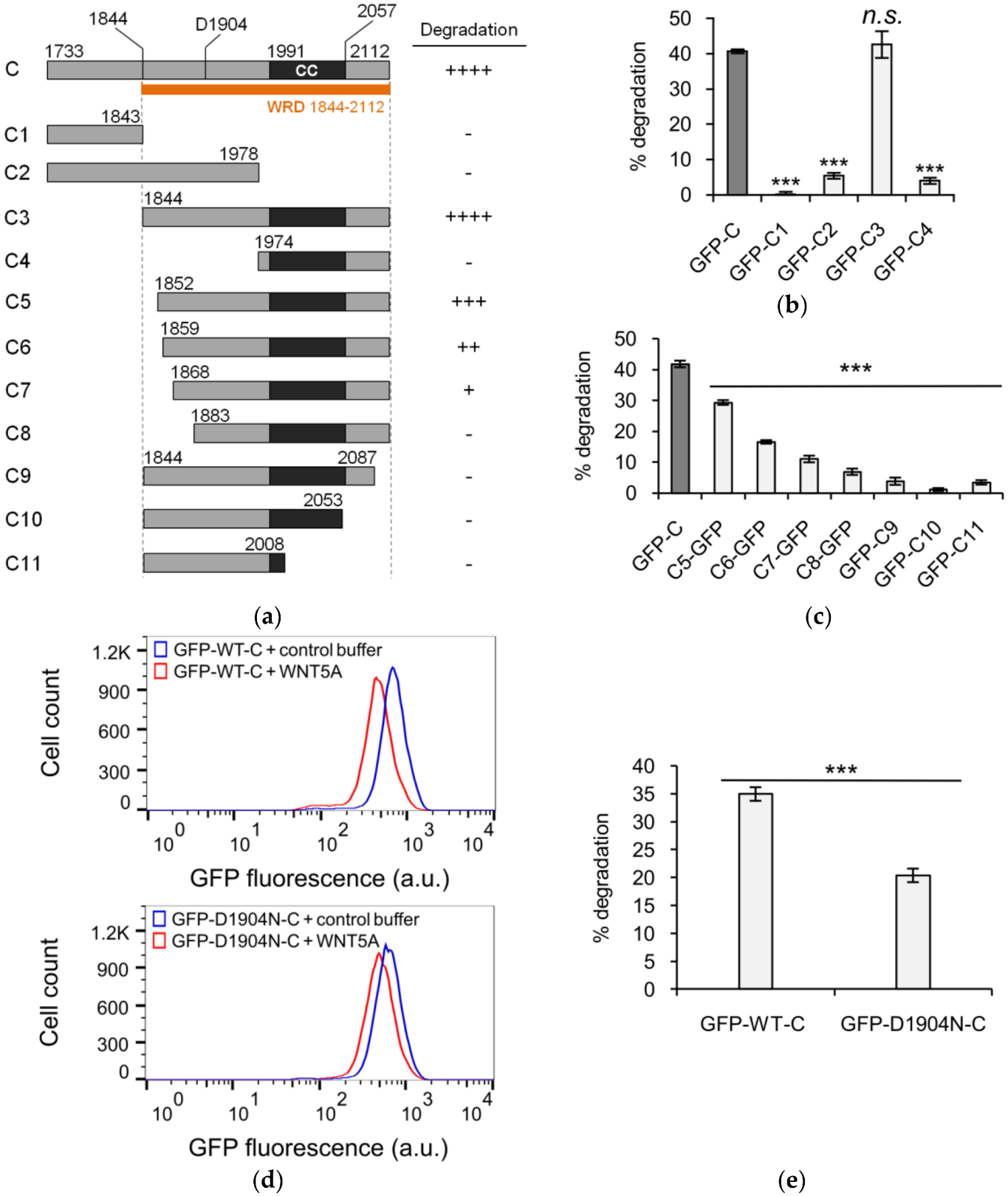
3.2. Defining the Sequence Elements within KIF26B-C that are Essential for WNT5A-Dependent Degradation
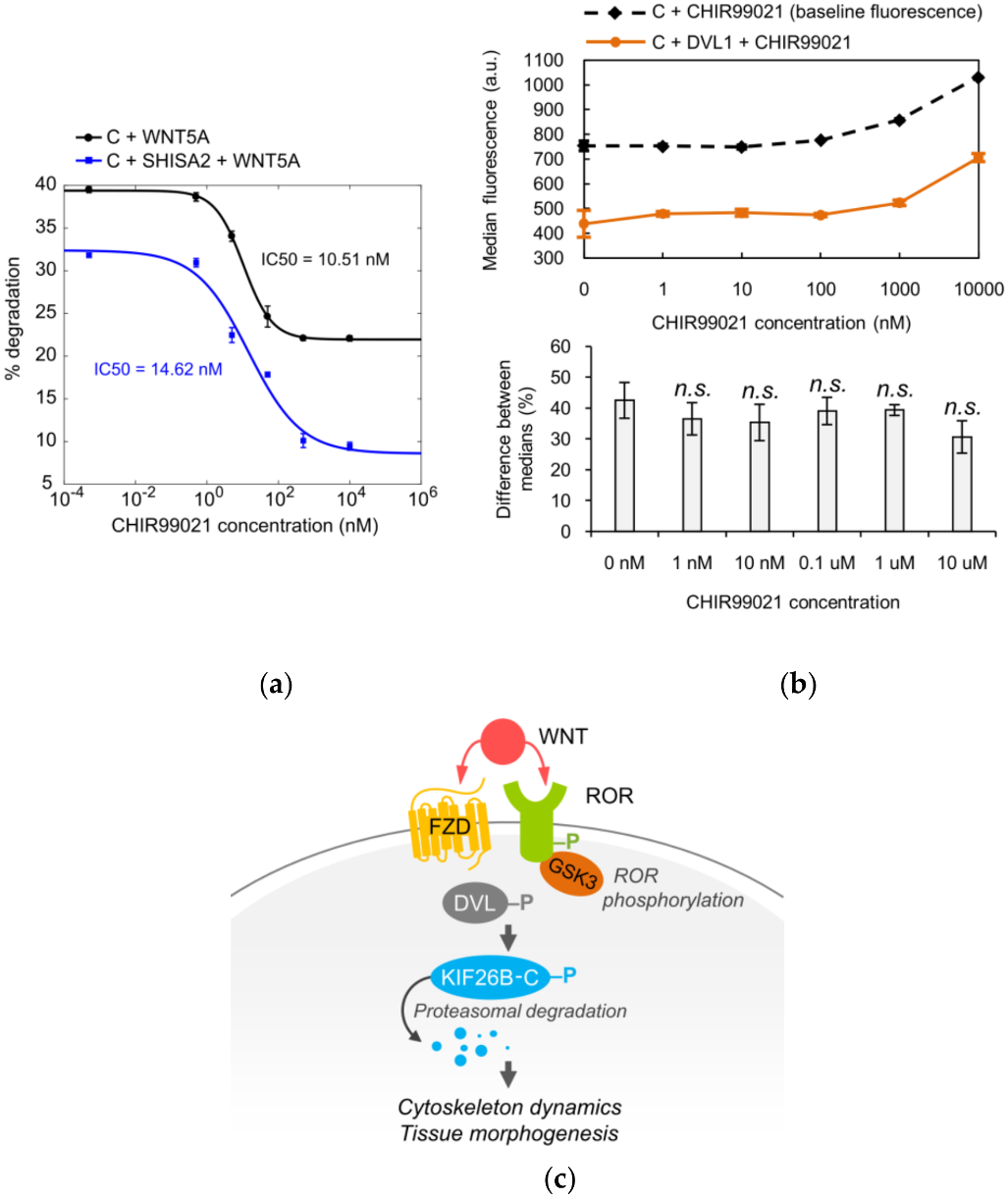
3.3. The Role of GSK3 in WNT5A Regulation of KIF26B Degradation
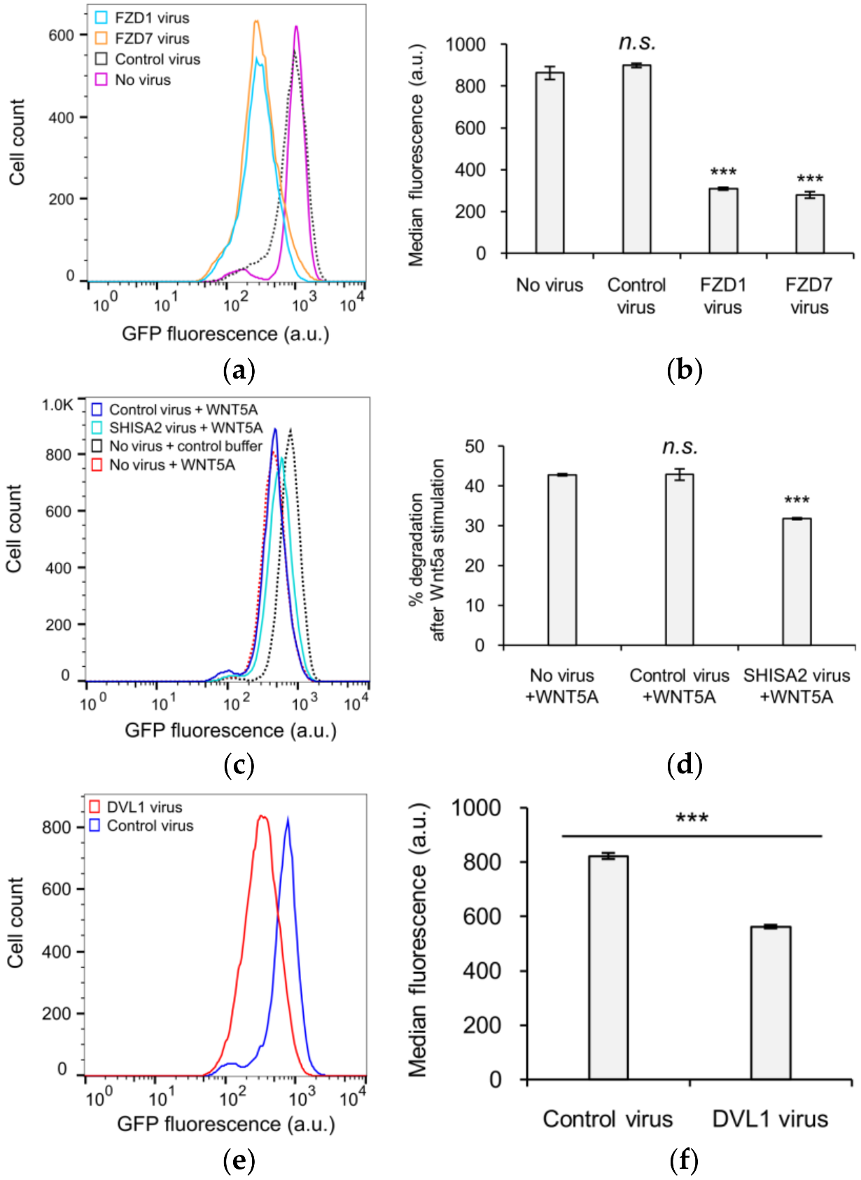
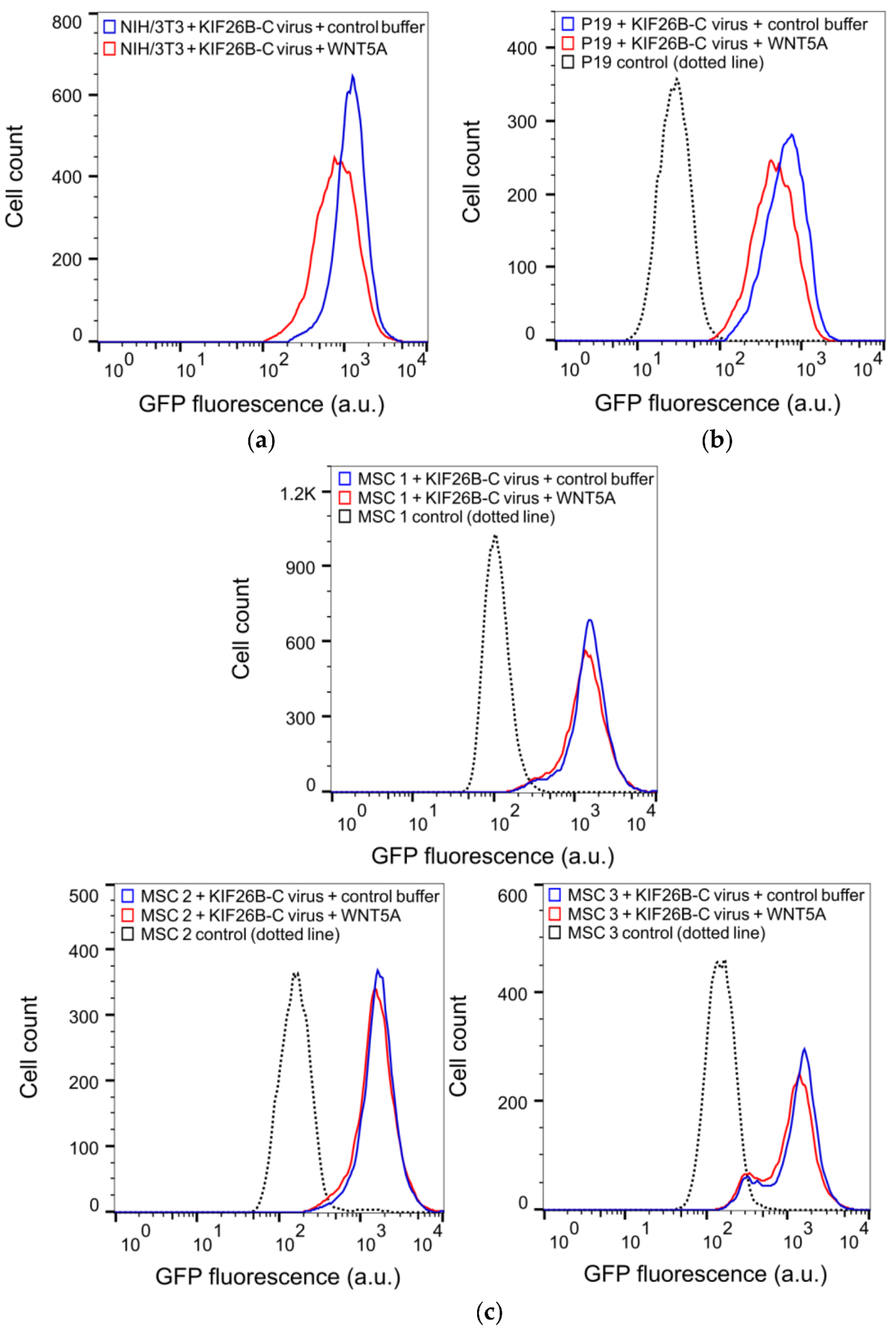
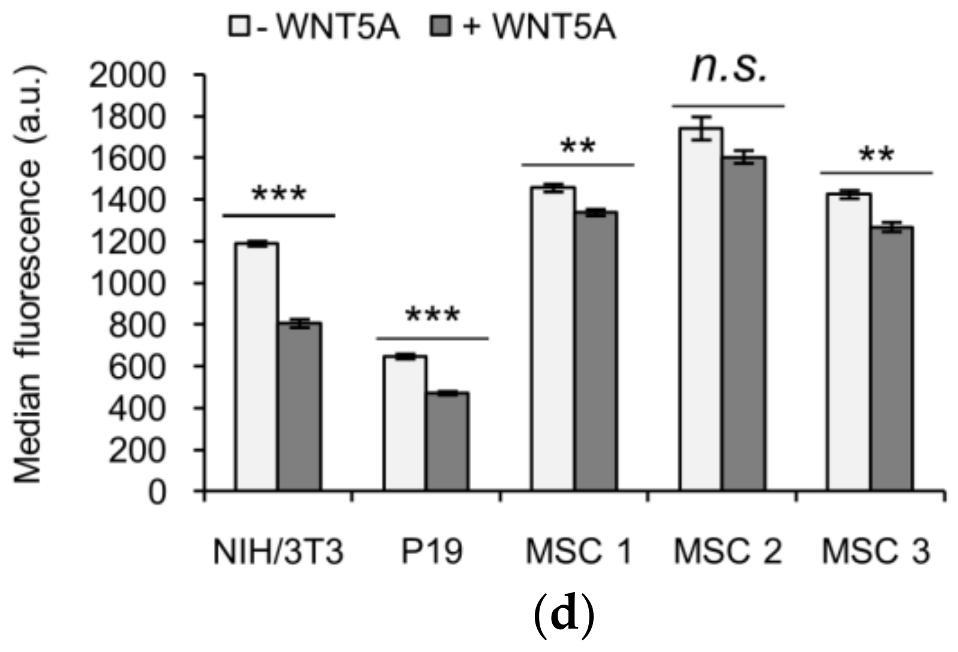
3.4. KIF26B-C as a Molecular Tool for Profiling Noncanonical WNT5A Signaling in Somatic and Stem Cells
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant Name | Forward/Reverse | Primer Sequence (5′→3′) |
|---|---|---|
| KIF26B-Delta N | F | gatcggccggcctaccatgcggaagaagcagatcacc |
| KIF26B-Delta N | R | gatcggcgcgccttatcggcgcctggaggtgatgtc |
| KIF26B-Delta M | F | gatcggccggcctaccatgaagaccaagtacacatcaa |
| KIF26B-Delta M | R | gatcggcgcgccttatcggcgcctggaggtgatgtc |
| KIF26B-Delta C | F | gatcggccggcctaccatgaattcggtagccggaaataaag |
| KIF26B-Delta C | R | gatcggcgcgccttacttgctcactgcagagatctt |
| KIF26B-C | F | gatcggccggcctaccatgtctaagatctctgcagtga |
| KIF26B-C | R | gatcggcgcgccttatcggcgcctggaggtgatgtc |
| KIF26B-C1 | F | gacttgaattcaggccggcctaccatgtctaagatctctg |
| KIF26B-C1 | R | tatagttctagaggcgcgccttatcggcgcctggaggtgatgtc |
| KIF26B-C2 | F | gacttgaattcaggccggcctaccatgtctaagatctctg |
| KIF26B-C2 | R | tatagttctagaggcgcgccttagccgtccacccagcggac |
| KIF26B-C3 | F | gacttgaattcaggccggcctaccaagcccgcagccgcccac |
| KIF26B-C3 | R | tatagttctagaggcgcgccttatcggcgcctggaggtgatgtc |
| KIF26B-C4 | F | gacttgaattcaggccggcctacccgctgggtggacggc |
| KIF26B-C4 | R | tatagttctagaggcgcgccttatcggcgcctggaggtgatgtc |
| KIF26B-C5 | F | gccggaaccaattcagtcgacaccatgccctcgccctacagcaag |
| KIF26B-C5 | R | ccttgctcaccatggttgtggcgcctcggcgcctggaggtgatg |
| KIF26B-C6 | F | gccggaaccaattcagtcgacaccatgacccctccgaggaagccg |
| KIF26B-C6 | R | ccttgctcaccatggttgtggcgcctcggcgcctggaggtgatg |
| KIF26B-C7 | F | gccggaaccaattcagtcgacaccatgagcagcgggcacggtagt |
| KIF26B-C7 | R | ccttgctcaccatggttgtggcgcctcggcgcctggaggtgatg |
| KIF26B-C8 | F | gccggaaccaattcagtcgacaccatgctaccacctgccatggg |
| KIF26B-C8 | R | ccttgctcaccatggttgtggcgcctcggcgcctggaggtgatg |
| KIF26B-C9 | F | gacttgaattcaggccggcctaccaagcccgcagccgcccac |
| KIF26B-C9 | R | gatcggcgcgccttacaagcgctctgtcacacc |
| KIF26B-C10 | F | gacttgaattcaggccggcctaccaagcccgcagccgcccac |
| KIF26B-C10 | R | gatcggcgcgccttacaggtactgcttggtcgc |
| KIF26B-C11 | F | gacttgaattcaggccggcctaccaagcccgcagccgcccac |
| KIF26B-C11 | R | gatcggcgcgccttatcgacgtcgctgcaggcg |
| KIF26B-C (D1904N) | F | ggctacgagagcatgatgagaaacagcgaggccaccggcagtg |
| KIF26B-C (D1904N) | R | cactgccggtggcctcgctgtttctcatcatgctctcgtagcc |
| KIF26A-C | F | gatcggccggcctaccatgagcccagccaagggtgttggag |
| KIF26A-C | R | gatcggcgcgcctcaaacatccacctcttgtggccc |
References
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Hoppler, S.; Moon, R.T. Wnt Signaling in Development and Disease: Molecular Mechanisms and Biological Functions; Wiley Blackwell: Hoboken, NJ, USA, 2014; 459p. [Google Scholar]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Veeman, M.T.; Axelrod, J.D.; Moon, R.T. A second canon. Functions and mechanisms of β-catenin-independent Wnt signaling. Dev. Cell 2003, 5, 367–377. [Google Scholar] [CrossRef]
- Carvallo, L.; Munoz, R.; Bustos, F.; Escobedo, N.; Carrasco, H.; Olivares, G.; Larrain, J. Non-canonical Wnt signaling induces ubiquitination and degradation of Syndecan4. J. Biol. Chem. 2010, 285, 29546–29555. [Google Scholar] [CrossRef] [PubMed]
- Kurayoshi, M.; Oue, N.; Yamamoto, H.; Kishida, M.; Inoue, A.; Asahara, T.; Yasui, W.; Kikuchi, A. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res. 2006, 66, 10439–10448. [Google Scholar] [CrossRef] [PubMed]
- Iioka, H.; Iemura, S.; Natsume, T.; Kinoshita, N. Wnt signalling regulates paxillin ubiquitination essential for mesodermal cell motility. Nat. Cell Biol. 2007, 9, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Susman, M.W.; Karuna, E.P.; Kunz, R.C.; Gujral, T.S.; Cantu, A.V.; Choi, S.S.; Jong, B.Y.; Okada, K.; Scales, M.K.; Hum, J.; et al. Kinesin superfamily protein Kif26b links Wnt5a-Ror signaling to the control of cell and tissue behaviors in vertebrates. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, Y.; Sakaguchi, M.; Terabayashi, T.; Inenaga, T.; Inoue, S.; Kobayashi, C.; Oshima, N.; Kiyonari, H.; Nakagata, N.; Sato, Y.; et al. Kif26b, a kinesin family gene, regulates adhesion of the embryonic kidney mesenchyme. Proc. Natl. Acad. Sci. USA 2010, 107, 9240–9245. [Google Scholar] [CrossRef] [PubMed]
- Guillabert-Gourgues, A.; Jaspard-Vinassa, B.; Bats, M.L.; Sewduth, R.N.; Franzl, N.; Peghaire, C.; Jeanningros, S.; Moreau, C.; Roux, E.; Larrieu-Lahargue, F.; et al. Kif26b controls endothelial cell polarity through the Dishevelled/Daam1-dependent planar cell polarity-signaling pathway. Mol. Biol. Cell 2016, 27, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Nibbeling, E.A.R.; Duarri, A.; Verschuuren-Bemelmans, C.C.; Fokkens, M.R.; Karjalainen, J.M.; Smeets, C.; de Boer-Bergsma, J.J.; van der Vries, G.; Dooijes, D.; Bampi, G.B.; et al. Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain 2017, 140, 2860–2878. [Google Scholar] [CrossRef] [PubMed]
- Fierro, F.A.; Kalomoiris, S.; Sondergaard, C.S.; Nolta, J.A. Effects on proliferation and differentiation of multipotent bone marrow stromal cells engineered to express growth factors for combined cell and gene therapy. Stem Cells 2011, 29, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Karuna, E.P.; Susman, M.W.; Ho, H.H. Quantitative live-cell reporter assay for noncanonical Wnt activity. Bio Protoc. 2018, 8. [Google Scholar] [CrossRef]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [PubMed]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.; Minneci, F.; Nugent, T.C.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 2013, 41, W349–W357. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A.; Van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Tanaka, Y. Kinesin superfamily proteins (KIFs): Various functions and their relevance for important phenomena in life and diseases. Exp. Cell Res. 2015, 334, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Oishi, I.; Suzuki, H.; Onishi, N.; Takada, R.; Kani, S.; Ohkawara, B.; Koshida, I.; Suzuki, K.; Yamada, G.; Schwabe, G.C.; et al. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells 2003, 8, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Mikels, A.; Minami, Y.; Nusse, R. Ror2 receptor requires tyrosine kinase activity to mediate Wnt5a signaling. J. Biol. Chem. 2009, 284, 30167–30176. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.Y.; Susman, M.W.; Bikoff, J.B.; Ryu, Y.K.; Jonas, A.M.; Hu, L.; Kuruvilla, R.; Greenberg, M.E. Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 4044–4051. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Yoo, S.K.; Nishita, M.; Kikuchi, A.; Minami, Y. Wnt5a modulates glycogen synthase kinase 3 to induce phosphorylation of receptor tyrosine kinase Ror2. Genes Cells 2007, 12, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Grumolato, L.; Liu, G.; Mong, P.; Mudbhary, R.; Biswas, R.; Arroyave, R.; Vijayakumar, S.; Economides, A.N.; Aaronson, S.A. Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev. 2010, 24, 2517–2530. [Google Scholar] [CrossRef] [PubMed]
- Bryja, V.; Schulte, G.; Rawal, N.; Grahn, A.; Arenas, E. Wnt-5a induces Dishevelled phosphorylation and dopaminergic differentiation via a CK1-dependent mechanism. J. Cell Sci. 2007, 120, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Terabayashi, T.; Sakaguchi, M.; Shinmyozu, K.; Ohshima, T.; Johjima, A.; Ogura, T.; Miki, H.; Nishinakamura, R. Phosphorylation of Kif26b promotes its polyubiquitination and subsequent proteasomal degradation during kidney development. PLoS ONE 2012, 7, e39714. [Google Scholar] [CrossRef] [PubMed]
- Ring, D.B.; Johnson, K.W.; Henriksen, E.J.; Nuss, J.M.; Goff, D.; Kinnick, T.R.; Ma, S.T.; Reeder, J.W.; Samuels, I.; Slabiak, T.; et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 2003, 52, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Karuna, E.P.; Ho, H.H.; University of California, Davis, CA, USA. Unpublished data. 2018.
- McBurney, M.W.; Rogers, B.J. Isolation of male embryonal carcinoma cells and their chromosome replication patterns. Dev. Biol. 1982, 89, 503–508. [Google Scholar] [CrossRef]
- Van der Heyden, M.A.; Defize, L.H. Twenty one years of P19 cells: What an embryonal carcinoma cell line taught us about cardiomyocyte differentiation. Cardiovasc. Res. 2003, 58, 292–302. [Google Scholar] [CrossRef]
- Mahla, R.S. Stem cells applications in regenerative medicine and disease therapeutics. Int. J. Cell Biol. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, S.C.; Sutton, C.A.; Brady, K.; Salerno, A.; Katopodi, T.; Williams, R.L.; West, C.C.; Evseenko, D.; Wu, L.; Pang, S.; et al. The Wnt5a receptor, receptor tyrosine kinase-like orphan receptor 2, is a predictive cell surface marker of human mesenchymal stem cells with an enhanced capacity for chondrogenic differentiation. Stem Cells 2017, 35, 2280–2291. [Google Scholar] [CrossRef] [PubMed]
- Takiguchi, G.; Nishita, M.; Kurita, K.; Kakeji, Y.; Minami, Y. Wnt5a-Ror2 signaling in mesenchymal stem cells promotes proliferation of gastric cancer cells by activating CXCL16-CXCR6 axis. Cancer Sci. 2016, 107, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.X.; Liu, A.R.; Chen, S.; He, H.L.; Chen, Q.H.; Xu, J.Y.; Pan, C.; Yang, Y.; Guo, F.M.; Huang, Y.Z.; et al. The orphan receptor tyrosine kinase ROR2 facilitates MSCs to repair lung injury in ARDS animal model. Cell Transplant. 2016, 25, 1561–1574. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Wang, Z.; Xin, F.; Xin, H. Wnt5a is associated with the differentiation of bone marrow mesenchymal stem cells in vascular calcification by connecting with different receptors. Mol. Med. Rep. 2014, 10, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.X.; Liu, A.R.; He, H.L.; Chen, Q.H.; Yang, Y.; Guo, F.M.; Huang, Y.Z.; Liu, L.; Qiu, H.B. Stable genetic alterations of β-catenin and ROR2 regulate the Wnt pathway, affect the fate of MSCs. J. Cell Physiol. 2014, 229, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Xin, F.; Zhou, S.; Guan, S. The Wnt5a/Ror2 pathway is associated with determination of the differentiation fate of bone marrow mesenchymal stem cells in vascular calcification. Int. J. Mol. Med. 2013, 31, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Sonomoto, K.; Yamaoka, K.; Oshita, K.; Fukuyo, S.; Zhang, X.; Nakano, K.; Okada, Y.; Tanaka, Y. Interleukin-1β induces differentiation of human mesenchymal stem cells into osteoblasts via the Wnt-5a/receptor tyrosine kinase-like orphan receptor 2 pathway. Arthritis Rheumatol. 2012, 64, 3355–3363. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Oishi, I.; Endo, M.; Nishita, M. Ror-family receptor tyrosine kinases in noncanonical Wnt signaling: Their implications in developmental morphogenesis and human diseases. Dev. Dyn. 2010, 239, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Ajani, J.A.; Hawk, E.T.; Ye, Y.; Lee, J.H.; Bhutani, M.S.; Hofstetter, W.L.; Swisher, S.G.; Wang, K.K.; Wu, X. Genome-wide catalogue of chromosomal aberrations in barrett’s esophagus and esophageal adenocarcinoma: A high-density single nucleotide polymorphism array analysis. Cancer Prev. Res. 2010, 3, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhao, Z.B.; Wang, G.; Hui, Z.; Wang, M.H.; Pan, J.F.; Zheng, H. High expression of KIF26B in breast cancer associates with poor prognosis. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Wang, J.; Cui, F.; Wang, X.; Xue, Y.; Chen, J.; Yu, Y.; Lu, H.; Zhang, M.; Tang, H.; Peng, Z. Elevated kinesin family member 26B is a prognostic biomarker and a potential therapeutic target for colorectal cancer. J. Exp. Clin. Cancer Res. 2015, 34. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T.; Furuichi, K.; Higashide, T.; Sakurai, M.; Hashimoto, S.; Shinozaki, Y.; Hara, A.; Iwata, Y.; Sakai, N.; Sugiyama, K.; et al. Association of PAX2 and other gene mutations with the clinical manifestations of renal coloboma syndrome. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Raun, N.; Mailo, J.; Spinelli, E.; He, X.; McAvena, S.; Brand, L.; O’Sullivan, J.; Andersen, J.; Richer, L.; Tang-Wai, R.; et al. Quantitative phenotypic and network analysis of 1q44 microdeletion for microcephaly. Am. J. Med. Genet. A 2017, 173, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.; Sim, S.H.; Lee, Y.; Moon, K.C.; Lee, C.; Park, W.Y.; Kim, N.K.; Lee, S.H.; Lee, H. Comprehensive somatic genome alterations of urachal carcinoma. J. Med. Genet. 2017, 54, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, R.R.; Wang, X.J.; Su, Z.X.; Chen, X.; Shi, D.B.; Guo, X.Y.; Liu, H.T.; Gao, P. KIF26B, a novel oncogene, promotes proliferation and metastasis by activating the VEGF pathway in gastric cancer. Oncogene 2017, 36, 5609–5619. [Google Scholar] [CrossRef] [PubMed]
- Marikawa, Y.; Tamashiro, D.A.; Fujita, T.C.; Alarcon, V.B. Aggregated P19 mouse embryonal carcinoma cells as a simple in vitro model to study the molecular regulations of mesoderm formation and axial elongation morphogenesis. Genesis 2009, 47, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Bilkovski, R.; Schulte, D.M.; Oberhauser, F.; Gomolka, M.; Udelhoven, M.; Hettich, M.M.; Roth, B.; Heidenreich, A.; Gutschow, C.; Krone, W.; et al. Role of WNT-5a in the determination of human mesenchymal stem cells into preadipocytes. J. Biol. Chem. 2010, 285, 6170–6178. [Google Scholar] [CrossRef] [PubMed]
- Baksh, D.; Boland, G.M.; Tuan, R.S. Cross-talk between Wnt signaling pathways in human mesenchymal stem cells leads to functional antagonism during osteogenic differentiation. J. Cell. Biochem. 2007, 101, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Topol, L.; Lee, H.; Wu, J. Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation. Development 2003, 130, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Cho, H.H.; Park, J.S.; Jeong, H.S. Non-canonical WNT mediated neurogenic differentiation of human bone marrow-derived mesenchymal stem cells. Neurosci. Lett. 2017, 660, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Etheridge, S.L.; Spencer, G.J.; Heath, D.J.; Genever, P.G. Expression profiling and functional analysis of Wnt signaling mechanisms in mesenchymal stem cells. Stem Cells 2004, 22, 849–860. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karuna, E.P.; Choi, S.S.; Scales, M.K.; Hum, J.; Cohen, M.; Fierro, F.A.; Ho, H.-Y.H. Identification of a WNT5A-Responsive Degradation Domain in the Kinesin Superfamily Protein KIF26B. Genes 2018, 9, 196. https://doi.org/10.3390/genes9040196
Karuna EP, Choi SS, Scales MK, Hum J, Cohen M, Fierro FA, Ho H-YH. Identification of a WNT5A-Responsive Degradation Domain in the Kinesin Superfamily Protein KIF26B. Genes. 2018; 9(4):196. https://doi.org/10.3390/genes9040196
Chicago/Turabian StyleKaruna, Edith P., Shannon S. Choi, Michael K. Scales, Jennie Hum, Michael Cohen, Fernando A. Fierro, and Hsin-Yi Henry Ho. 2018. "Identification of a WNT5A-Responsive Degradation Domain in the Kinesin Superfamily Protein KIF26B" Genes 9, no. 4: 196. https://doi.org/10.3390/genes9040196
APA StyleKaruna, E. P., Choi, S. S., Scales, M. K., Hum, J., Cohen, M., Fierro, F. A., & Ho, H. -Y. H. (2018). Identification of a WNT5A-Responsive Degradation Domain in the Kinesin Superfamily Protein KIF26B. Genes, 9(4), 196. https://doi.org/10.3390/genes9040196