Wnt Signaling in Thyroid Homeostasis and Carcinogenesis


Abstract
:1. Introduction
2. Thyroid Physiology and the Molecular Mechanisms of Thyroid Hormone Function
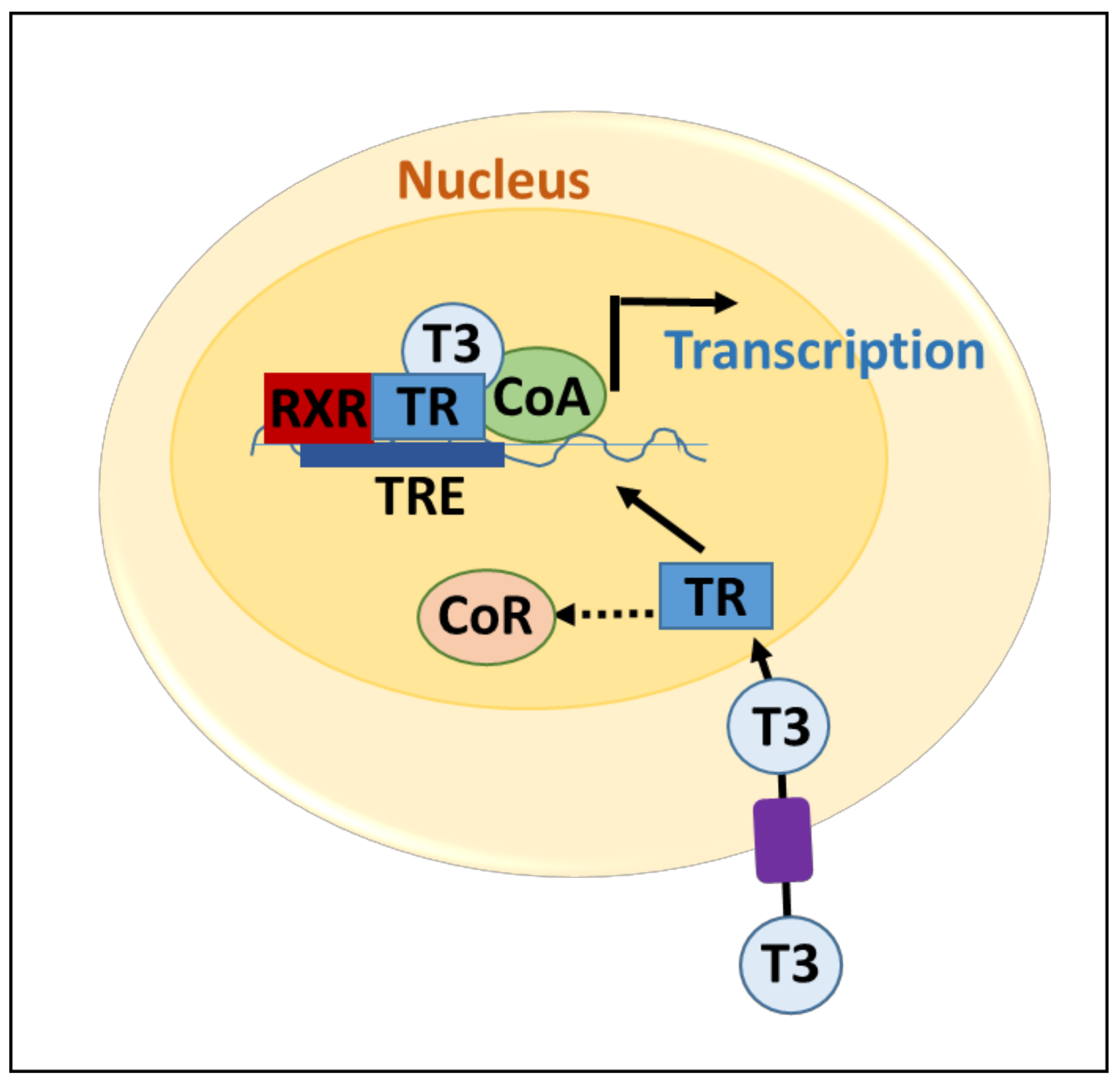
2.1. Thyroid Hormone Regulation and Signaling
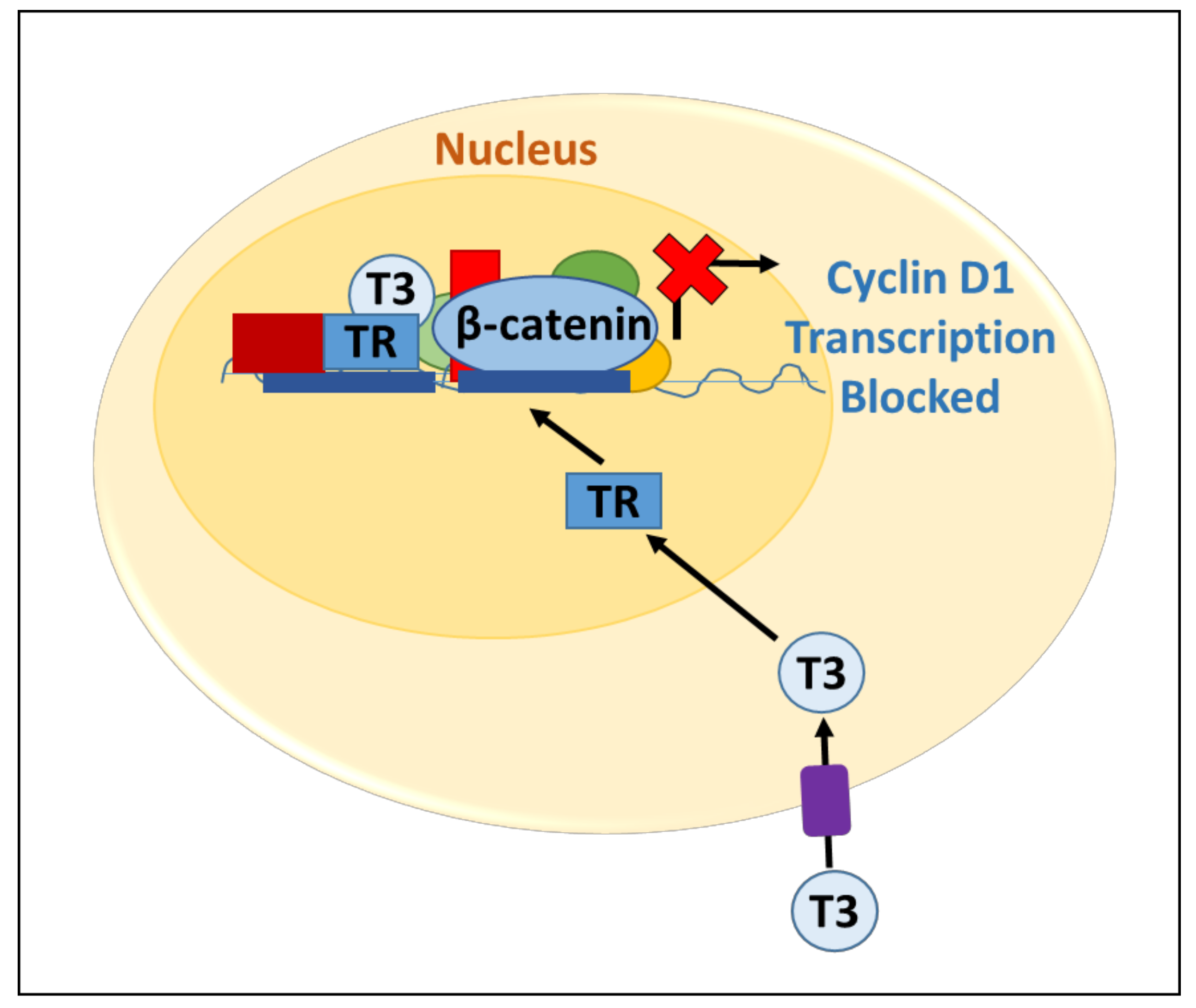
2.2. Regulation of the Wnt Pathway by Thyroid Hormone Receptors
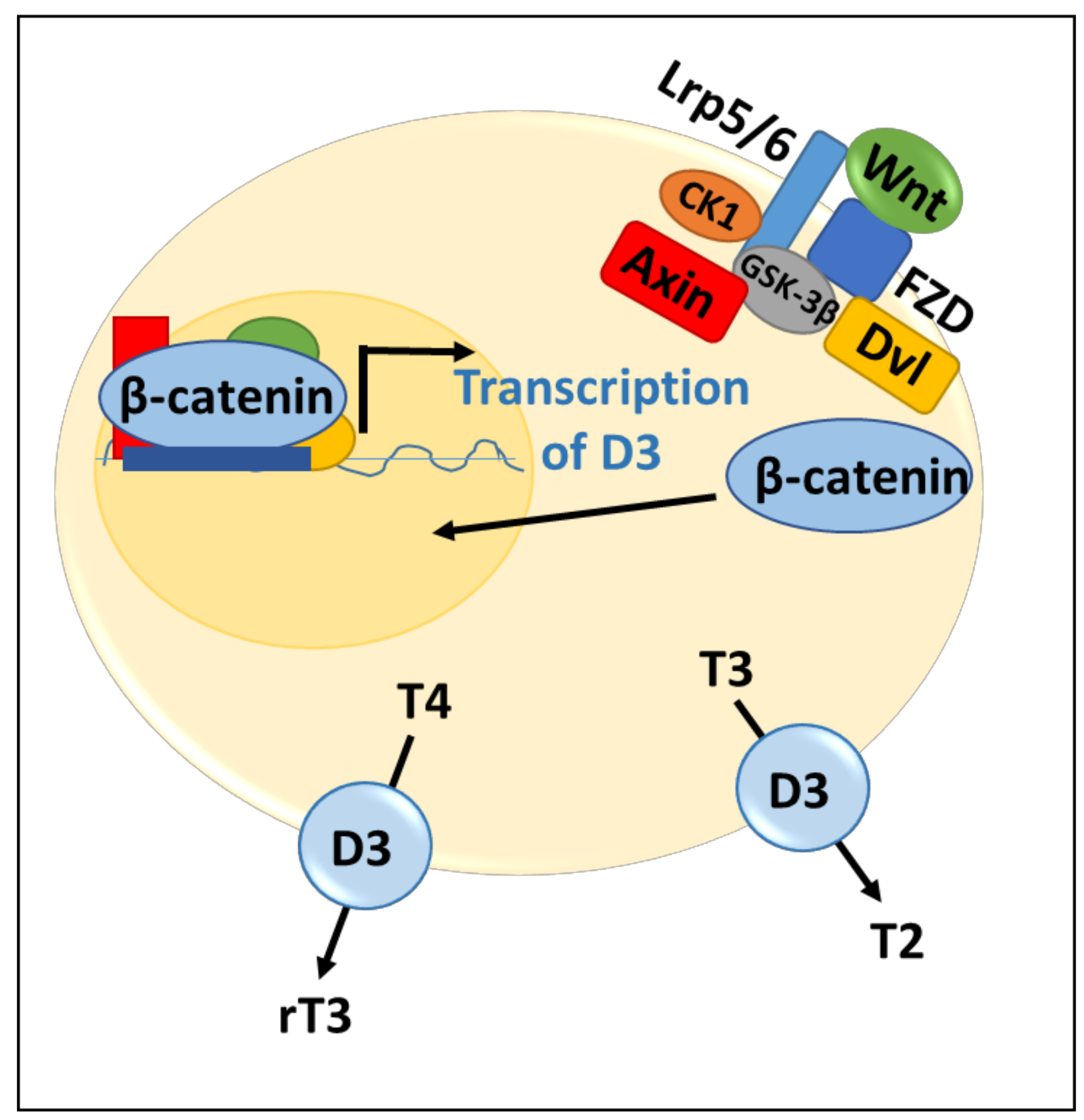
2.3. Control of Thyroid Hormone Function by the Wnt Pathway
3. Thyroid Gland Development and Adult Stem Cells
3.1. Thyroid Gland Development
3.2. Thyroid Gland Stem Cells
3.3. Wnt Signaling in Thyroid Stem Cells
4. Thyroid Cancer and the Role of Wnt Signaling
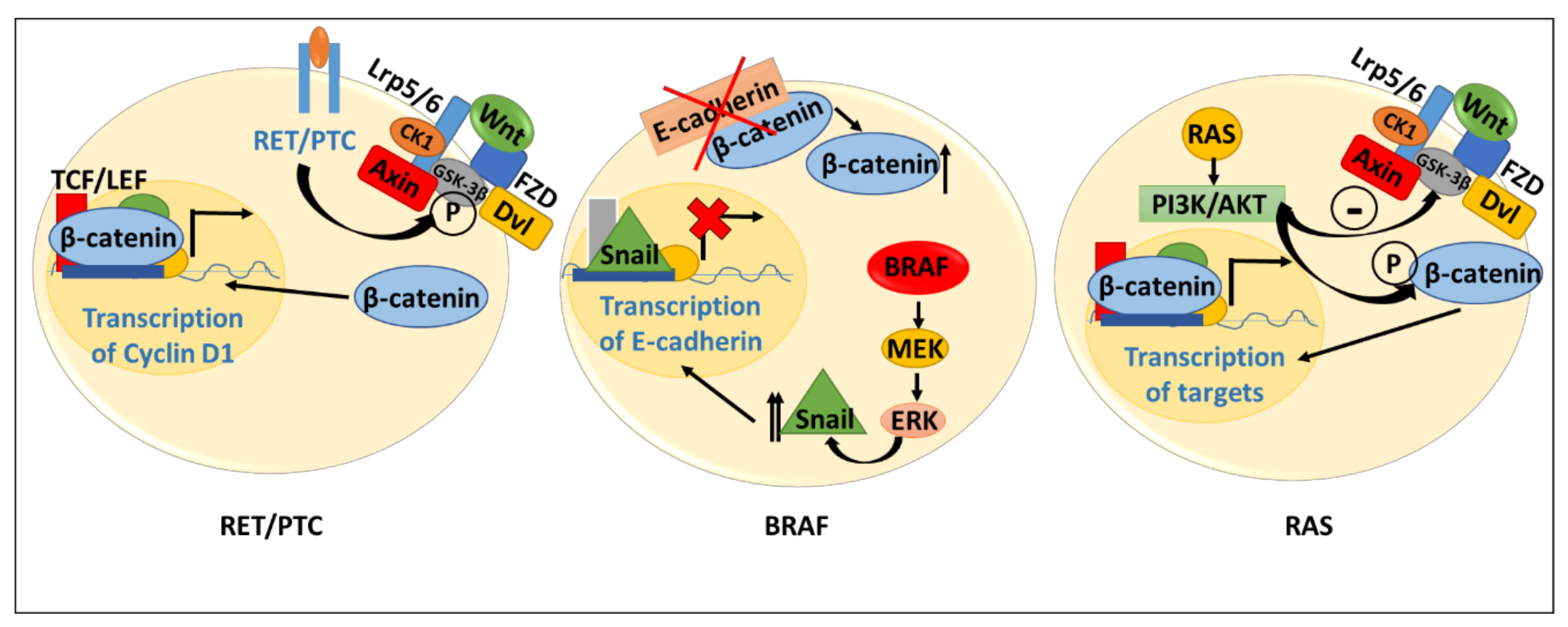
4.1. Evidence for Wnt Signaling in Thyroid Cancer Genesis
4.2. Evidence for Thyroid Cancer Stem Cells
4.3. Wnt Signaling in Thyroid Cancer Stem Cells
5. Conclusions
Conflicts of Interest
References
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5+ve stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef]
- Biteau, B.; Hochmuth, C.E.; Jasper, H. Maintaining tissue homeostasis: Dynamic control of somatic stem cell activity. Cell Stem Cell 2011, 9, 402–411. [Google Scholar] [CrossRef]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef]
- Fleming, H.E.; Janzen, V.; Lo Celso, C.; Guo, J.; Leahy, K.M.; Kronenberg, H.M.; Scadden, D.T. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell 2008, 2, 274–283. [Google Scholar] [CrossRef]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R., 3rd; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef]
- Sastre-Perona, A.; Santisteban, P. Role of the Wnt pathway in thyroid cancer. Front. Endocrinol. (Lausanne) 2012, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Sastre-Perona, A.; Santisteban, P. Wnt-independent role of β-catenin in thyroid cell proliferation and differentiation. Mol. Endocrinol. 2014, 28, 681–695. [Google Scholar] [CrossRef]
- Garber, J.R.; Cobin, R.H.; Gharib, H.; Hennessey, J.V.; Klein, I.; Mechanick, J.I.; Pessah-Pollack, R.; Singer, P.A.; Woeber, K.A. American Association of Clinical Endocrinologists and American Thyroid Association Taskforce on Hypothyroidism in Adults. Clinical practice guidelines for hypothyroidism in adults: Cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocr. Pract. 2012, 18, 988–1028. [Google Scholar]
- Yen, P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, B.; Koenig, R.J. Thyroid hormone response element sequence and the recruitment of retinoid X receptors for thyroid hormone responsiveness. J. Biol. Chem. 2001, 276, 3929–3936. [Google Scholar] [CrossRef]
- Dentice, M.; Marsili, A.; Zavacki, A.; Larsen, P.R.; Salvatore, D. The deiodinases and the control of intracellular thyroid hormone signaling during cellular differentiation. Biochim. Biophys. Acta 2013, 1830, 3937–3945. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Robson, H.; Kester, M.H.; van Leeuwen, J.P.; Shalet, S.M.; Visser, T.J.; Williams, G.R. Iodothyronine deiodinase enzyme activities in bone. Bone 2008, 43, 126–134. [Google Scholar] [CrossRef]
- Gereben, B.; Zeold, A.; Dentice, M.; Salvatore, D.; Bianco, A.C. Activation and inactivation of thyroid hormone by deiodinases: Local action with general consequences. Cell Mol. Life Sci. 2008, 65, 570–590. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Iovino, F.; Eterno, V.; Cammareri, P.; Gambara, G.; Espina, V.; Gulotta, G.; Dieli, F.; Giordano, S.; De Maria, R.; et al. Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res. 2010, 70, 8874–8885. [Google Scholar] [CrossRef] [PubMed]
- Guigon, C.J.; Zhao, L.; Lu, C.; Willingham, M.C.; Cheng, S.Y. Regulation of β-catenin by a novel nongenomic action of thyroid hormone β receptor. Mol. Cell Biol. 2008, 28, 4598–4608. [Google Scholar] [CrossRef]
- Dentice, M.; Luongo, C.; Ambrosio, R.; Sibilio, A.; Casillo, A.; Iaccarino, A.; Troncone, G.; Fenzi, G.; Larsen, P.R.; Salvatore, D. β-Catenin regulates deiodinase levels and thyroid hormone signaling in colon cancer cells. Gastroenterology 2012, 143, 1037–1047. [Google Scholar] [CrossRef]
- Brown, A.R.; Simmen, R.C.; Simmen, F.A. The role of thyroid hormone signaling in the prevention of digestive system cancers. Int. J. Mol. Sci. 2013, 14, 16240–16257. [Google Scholar] [CrossRef]
- Saito-Diaz, K.; Chen, T.W.; Wang, X.; Thorne, C.A.; Wallace, H.A.; Page-McCaw, A.; Lee, E. The way Wnt works: Components and mechanism. Growth Factors 2013, 31, 1–31. [Google Scholar] [CrossRef]
- Ng, L.; Lyubarsky, A.; Nikonov, S.S.; Ma, M.; Srinivas, M.; Kefas, B.; St Germain, D.L.; Hernandez, A.; Pugh, E.N., Jr.; Forrest, D. Type 3 deiodinase, a thyroid-hormone-inactivating enzyme, controls survival and maturation of cone photoreceptors. J. Neurosci. 2010, 30, 3347–3357. [Google Scholar] [CrossRef]
- Dentice, M.; Salvatore, D. Deiodinases: The balance of thyroid hormone: Local impact of thyroid hormone inactivation. J. Endocrinol. 2011, 209, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Dentice, M.; Antonini, D.; Salvatore, D. Type 3 deiodinase and solid tumors: An intriguing pair. Expert Opin. Ther. Targets 2013, 17, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.D.; Park, K.S.; Guo, Q.M.; Alkharouf, N.W.; Malek, R.L.; Lee, N.H.; Liu, E.T.; Cheng, S.Y. Silencing of Wnt signaling and activation of multiple metabolic pathways in response to thyroid hormone-stimulated cell proliferation. Mol. Cell Biol. 2001, 21, 6626–6639. [Google Scholar] [CrossRef] [PubMed]
- Sirakov, M.; Skah, S.; Nadjar, J.; Plateroti, M. Thyroid hormone’s action on progenitor/stem cell biology: New challenge for a classic hormone? Biochim. Biophys. Acta 2013, 1830, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Natsume, H.; Sasaki, S.; Kitagawa, M.; Kashiwabara, Y.; Matsushita, A.; Nakano, K.; Nishiyama, K.; Nagayama, K.; Misawa, H.; Masuda, H.; et al. β-catenin/Tcf-1-mediated transactivation of cyclin D1 promoter is negatively regulated by thyroid hormone. Biochem. Biophys. Res. Commun. 2003, 309, 408–413. [Google Scholar] [CrossRef]
- Bianco, A.C.; Kim, B.W. Deiodinases: Implications of the local control of thyroid hormone action. J. Clin. Investig. 2006, 116, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Heijlen, M.; Houbrechts, A.M.; Bagci, E.; Van Herck, S.L.; Kersseboom, S.; Esguerra, C.V.; Blust, R.; Visser, T.J.; Knapen, D.; Darras, V.M. Knockdown of type 3 iodothyronine deiodinase severely perturbs both embryonic and early larval development in zebrafish. Endocrinology 2014, 155, 1547–1559. [Google Scholar] [CrossRef]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef]
- Bienz, M.; Clevers, H. Linking colorectal cancer to Wnt signaling. Cell 2000, 103, 311–320. [Google Scholar] [CrossRef]
- Thomas, T.; Nowka, K.; Lan, L.; Derwahl, M. Expression of endoderm stem cell markers: Evidence for the presence of adult stem cells in human thyroid glands. Thyroid 2006, 16, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Kimura, H.; Landek-Salgado, M.A.; Hagedorn, J.; Kimura, M.; Suzuki, K.; Westra, W.; Rose, N.R.; Caturegli, P. Regenerative potentials of the murine thyroid in experimental autoimmune thyroiditis: Role of CD24. Endocrinology 2009, 150, 492–499. [Google Scholar] [CrossRef]
- Mauchamp, J.; Mirrione, A.; Alquier, C.; Andre, F. Follicle-like structure and polarized monolayer: Role of the extracellular matrix on thyroid cell organization in primary culture. Biol Cell 1998, 90, 369–380. [Google Scholar] [CrossRef]
- Davies, T.F.; Latif, R.; Minsky, N.C.; Ma, R. Clinical review: The emerging cell biology of thyroid stem cells. J. Clin. Endocrinol. Metab. 2011, 96, 2692–2702. [Google Scholar] [CrossRef]
- Nilsson, M.; Fagman, H. Development of the thyroid gland. Development 2017, 144, 2123–2140. [Google Scholar] [CrossRef] [PubMed]
- Postiglione, M.P.; Parlato, R.; Rodriguez-Mallon, A.; Rosica, A.; Mithbaokar, P.; Maresca, M.; Marians, R.C.; Davies, T.F.; Zannini, M.S.; De Felice, M.; et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc. Natl. Acad. Sci. USA 2002, 99, 15462–15467. [Google Scholar] [CrossRef] [PubMed]
- Marians, R.C.; Ng, L.; Blair, H.C.; Unger, P.; Graves, P.N.; Davies, T.F. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc. Natl. Acad. Sci. USA 2002, 99, 15776–15781. [Google Scholar] [CrossRef]
- Xu, P.X.; Zheng, W.; Laclef, C.; Maire, P.; Maas, R.L.; Peters, H.; Xu, X. Eya1 is required for the morphogenesis of mammalian thymus, parathyroid and thyroid. Development 2002, 129, 3033–3044. [Google Scholar]
- Kurihara, Y.; Kurihara, H.; Maemura, K.; Kuwaki, T.; Kumada, M.; Yazaki, Y. Impaired development of the thyroid and thymus in endothelin-1 knockout mice. J. Cardiovasc. Pharmacol. 1995, 26 (Suppl. 3), S13–S16. [Google Scholar] [CrossRef]
- Kurmann, A.A.; Serra, M.; Hawkins, F.; Rankin, S.A.; Mori, M.; Astapova, I.; Ullas, S.; Lin, S.; Bilodeau, M.; Rossant, J.; et al. Regeneration of thyroid function by transplantation of differentiated pluripotent stem cells. Cell Stem Cell 2015, 17, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Elsalini, O.A.; von Gartzen, J.; Cramer, M.; Rohr, K.B. Zebrafish hhex, nk2.1a, and pax2.1 regulate thyroid growth and differentiation downstream of Nodal-dependent transcription factors. Dev. Biol. 2003, 263, 67–80. [Google Scholar] [CrossRef]
- Meeus, L.; Gilbert, B.; Rydlewski, C.; Parma, J.; Roussie, A.L.; Abramowicz, M.; Vilain, C.; Christophe, D.; Costagliola, S.; Vassart, G. Characterization of a novel loss of function mutation of PAX8 in a familial case of congenital hypothyroidism with in-place, normal-sized thyroid. J. Clin. Endocrinol. Metab. 2004, 89, 4285–4291. [Google Scholar] [CrossRef]
- De Felice, M.; Ovitt, C.; Biffali, E.; Rodriguez-Mallon, A.; Arra, C.; Anastassiadis, K.; Macchia, P.E.; Mattei, M.G.; Mariano, A.; Scholer, H.; et al. A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat. Genet. 1998, 19, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Clifton-Bligh, R.J.; Wentworth, J.M.; Heinz, P.; Crisp, M.S.; John, R.; Lazarus, J.H.; Ludgate, M.; Chatterjee, V.K. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat. Genet. 1998, 19, 399–401. [Google Scholar] [CrossRef]
- Antonica, F.; Kasprzyk, D.F.; Opitz, R.; Iacovino, M.; Liao, X.H.; Dumitrescu, A.M.; Refetoff, S.; Peremans, K.; Manto, M.; Kyba, M.; et al. Generation of functional thyroid from embryonic stem cells. Nature 2012, 491, 66–71. [Google Scholar] [CrossRef]
- Dumont, J.E.; Lamy, F.; Roger, P.; Maenhaut, C. Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiol. Rev. 1992, 72, 667–697. [Google Scholar] [CrossRef]
- Watanabe, H.; Gould, M.N.; Mahler, P.A.; Mulcahy, R.T.; Clifton, K.H. The influence of donor and recipient age and sex on the quantitative transplantation of monodispersed rat thyroid cells. Endocrinology 1983, 112, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, N.; Iwao, A.; Nagai, K.; Namba, H.; Ohtsuru, A.; Saenko, V.; Yamashita, S. Characterization of side population in thyroid cancer cell lines: Cancer stem-like cells are enriched partly but not exclusively. Endocrinology 2007, 148, 1797–1803. [Google Scholar] [CrossRef]
- Helmbrecht, K.; Kispert, A.; von Wasielewski, R.; Brabant, G. Identification of a Wnt/β-catenin signaling pathway in human thyroid cells. Endocrinology 2001, 142, 5261–5266. [Google Scholar] [CrossRef]
- Chen, G.; Jiang, Q.; You, Z.; Yao, J.; Mou, L.; Lin, X.; Shen, X.; You, T.; Lin, Q.; Wen, J.; et al. Regulation of GSK-3 beta in the proliferation and apoptosis of human thyrocytes investigated using a GSK-3 beta-targeting RNAi adenovirus expression vector: Involvement the Wnt/beta-catenin pathway. Mol. Biol. Rep. 2010, 37, 2773–2779. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Soravia, C.; Sugg, S.L.; Berk, T.; Mitri, A.; Cheng, H.; Gallinger, S.; Cohen, Z.; Asa, S.L.; Bapat, B.V. Familial adenomatous polyposis-associated thyroid cancer: A clinical, pathological, and molecular genetics study. Am. J. Pathol. 1999, 154, 127–135. [Google Scholar] [CrossRef]
- Kurihara, K.; Shimizu, S.; Chong, J.; Hishima, T.; Funata, N.; Kashiwagi, H.; Nagai, H.; Miyaki, M.; Fukayama, M. Nuclear localization of immunoreactive β-catenin is specific to familial adenomatous polyposis in papillary thyroid carcinoma. Jpn. J. Cancer Res. 2000, 91, 1100–1102. [Google Scholar] [CrossRef]
- Xu, B.; Yoshimoto, K.; Miyauchi, A.; Kuma, S.; Mizusawa, N.; Hirokawa, M.; Sano, T. Cribriform-morular variant of papillary thyroid carcinoma: A pathological and molecular genetic study with evidence of frequent somatic mutations in exon 3 of the β-catenin gene. J. Pathol. 2003, 199, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Cetta, F.; Chiappetta, G.; Melillo, R.M.; Petracci, M.; Montalto, G.; Santoro, M.; Fusco, A. The ret/ptc1 oncogene is activated in familial adenomatous polyposis-associated thyroid papillary carcinomas. J. Clin. Endocrinol. Metab. 1998, 83, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Cetta, F.; Montalto, G.; Gori, M.; Curia, M.C.; Cama, A.; Olschwang, S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: Results from a European cooperative study. J. Clin. Endocrinol. Metab. 2000, 85, 286–292. [Google Scholar] [PubMed]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E. RET/PTC rearrangement in thyroid tumors. Endocr. Pathol. 2002, 13, 3–16. [Google Scholar] [CrossRef]
- Cassinelli, G.; Favini, E.; Degl’Innocenti, D.; Salvi, A.; De Petro, G.; Pierotti, M.A.; Zunino, F.; Borrello, M.G.; Lanzi, C. RET/PTC1-driven neoplastic transformation and proinvasive phenotype of human thyrocytes involve Met induction and β-catenin nuclear translocation. Neoplasia 2009, 11, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Tartari, C.J.; Donadoni, C.; Manieri, E.; Mologni, L.; Mina, P.D.; Villa, A.; Gambacorti-Passerini, C. Dissection of the RET/β-catenin interaction in the TPC1 thyroid cancer cell line. Am. J. Cancer Res. 2011, 1, 716–725. [Google Scholar] [PubMed]
- Dong, W.; Zhang, H.; Li, J.; Guan, H.; He, L.; Wang, Z.; Shan, Z.; Teng, W. Estrogen induces metastatic potential of papillary thyroid cancer cells through estrogen receptor α and β. Int. J. Endocrinol. 2013, 2013, 941568. [Google Scholar] [CrossRef] [PubMed]
- Castellone, M.D.; De Falco, V.; Rao, D.M.; Bellelli, R.; Muthu, M.; Basolo, F.; Fusco, A.; Gutkind, J.S.; Santoro, M. The β-catenin axis integrates multiple signals downstream from RET/papillary thyroid carcinoma leading to cell proliferation. Cancer Res. 2009, 69, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Bikkavilli, R.K.; Malbon, C.C. Mitogen-activated protein kinases and Wnt/β-catenin signaling: Molecular conversations among signaling pathways. Commun. Integr. Biol. 2009, 2, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Juan, J.; Muraguchi, T.; Iezza, G.; Sears, R.C.; McMahon, M. Diminished WNT -> β-catenin -> c-MYC signaling is a barrier for malignant progression of BRAFV600E-induced lung tumors. Genes Dev. 2014, 28, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.E.; Curley, D.P.; Santhanakrishnan, M.; Rosenbaum, L.E.; Platt, J.T.; Gould Rothberg, B.E.; Taketo, M.M.; Dankort, D.; Rimm, D.L.; McMahon, M.; et al. β-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 2011, 20, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Baquero, P.; Sanchez-Hernandez, I.; Jimenez-Mora, E.; Orgaz, J.L.; Jimenez, B.; Chiloeches, A. V600EBRAF promotes invasiveness of thyroid cancer cells by decreasing E-cadherin expression through a Snail-dependent mechanism. Cancer Lett. 2013, 335, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Yi, J.W.; Park, C.H.; Lim, Y.; Lee, K.H.; Lee, K.E.; Kim, J.H. Role of BRAF and RAS mutations in extrathyroidal extension in papillary thyroid cancer. Cancer Genom. Proteom. 2016, 13, 171–181. [Google Scholar]
- De Menna, M.; D’Amato, V.; Ferraro, A.; Fusco, A.; Di Lauro, R.; Garbi, C.; De Vita, G. Wnt4 inhibits cell motility induced by oncogenic Ras. Oncogene 2013, 32, 4110–4119. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Perona, A.; Riesco-Eizaguirre, G.; Zaballos, M.A.; Santisteban, P. β-catenin signaling is required for RAS-driven thyroid cancer through PI3K activation. Oncotarget 2016, 7, 49435–49449. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Lee, E.J.; Kim, H.; Kim, S.H.; Ahn, H.Y.; Kim, Y.A.; Yi, K.H.; Park, D.J.; Shin, C.S.; Ahn, S.H.; et al. Dickkopf-1 inhibits thyroid cancer cell survival and migration through regulation of β-catenin/E-cadherin signaling. Mol. Cell Endocrinol. 2013, 366, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Furuya, F.; Hanover, J.A.; Cheng, S.Y. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone β receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 1780–1785. [Google Scholar] [CrossRef]
- Lu, C.; Zhu, X.; Willingham, M.C.; Cheng, S.Y. Activation of tumor cell proliferation by thyroid hormone in a mouse model of follicular thyroid carcinoma. Oncogene 2012, 31, 2007–2016. [Google Scholar] [CrossRef]
- Bergh, J.J.; Lin, H.Y.; Lansing, L.; Mohamed, S.N.; Davis, F.B.; Mousa, S.; Davis, P.J. Integrin αVβ3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology 2005, 146, 2864–2871. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Camp, R.L.; Herrero, A.; Carcangiu, M.L.; Rimm, D.L.; Tallini, G. β-catenin dysregulation in thyroid neoplasms: Down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am. J. Pathol. 2001, 158, 987–996. [Google Scholar] [CrossRef]
- Kurihara, T.; Ikeda, S.; Ishizaki, Y.; Fujimori, M.; Tokumoto, N.; Hirata, Y.; Ozaki, S.; Okajima, M.; Sugino, K.; Asahara, T. Immunohistochemical and sequencing analyses of the Wnt signaling components in Japanese anaplastic thyroid cancers. Thyroid 2004, 14, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Cali, G.; Gentile, F.; Mogavero, S.; Pallante, P.; Nitsch, R.; Ciancia, G.; Ferraro, A.; Fusco, A.; Nitsch, L. CDH16/Ksp-cadherin is expressed in the developing thyroid gland and is strongly down-regulated in thyroid carcinomas. Endocrinology 2012, 153, 522–534. [Google Scholar] [CrossRef]
- Brabant, G.; Hoang-Vu, C.; Cetin, Y.; Dralle, H.; Scheumann, G.; Molne, J.; Hansson, G.; Jansson, S.; Ericson, L.E.; Nilsson, M. E-cadherin: A differentiation marker in thyroid malignancies. Cancer Res. 1993, 53, 4987–4993. [Google Scholar]
- Rao, A.S.; Kremenevskaja, N.; von Wasielewski, R.; Jakubcakova, V.; Kant, S.; Resch, J.; Brabant, G. Wnt/β-catenin signaling mediates antineoplastic effects of imatinib mesylate (Gleevec) in anaplastic thyroid cancer. J. Clin. Endocrinol. Metab. 2006, 91, 159–168. [Google Scholar] [CrossRef]
- Du, S.J.; Purcell, S.M.; Christian, J.L.; McGrew, L.L.; Moon, R.T. Identification of distinct classes and functional domains of Wnts through expression of wild-type and chimeric proteins in Xenopus embryos. Mol. Cell Biol. 1995, 15, 2625–2634. [Google Scholar] [CrossRef]
- Kremenevskaja, N.; von Wasielewski, R.; Rao, A.S.; Schofl, C.; Andersson, T.; Brabant, G. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 2005, 24, 2144–2154. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E. Thyroid carcinoma: Molecular pathways and therapeutic targets. Mod. Pathol. 2008, 21 (Suppl. 2), S37–S43. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, K.; Namba, H.; Nakashima, M.; Nakayama, T.; Mitsutake, N.; Hayashi, T.; Maeda, S.; Ichinose, M.; Kanematsu, T.; Yamashita, S. Aberrant localization of β-catenin correlates with overexpression of its target gene in human papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2002, 87, 3433–3440. [Google Scholar] [PubMed]
- Rezk, S.; Brynes, R.K.; Nelson, V.; Thein, M.; Patwardhan, N.; Fischer, A.; Khan, A. β-Catenin expression in thyroid follicular lesions: Potential role in nuclear envelope changes in papillary carcinomas. Endocr. Pathol. 2004, 15, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Farrow, B.; Evers, B.M. Activation of PPARγ increases PTEN expression in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2003, 301, 50–53. [Google Scholar] [CrossRef]
- Lan, L.; Luo, Y.; Cui, D.; Shi, B.Y.; Deng, W.; Huo, L.L.; Chen, H.L.; Zhang, G.Y.; Deng, L.L. Epithelial-mesenchymal transition triggers cancer stem cell generation in human thyroid cancer cells. Int. J. Oncol. 2013, 43, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Takano, T. Fetal cell carcinogenesis of the thyroid: A modified theory based on recent evidence. Endocr. J. 2014, 61, 311–320. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Preto, A.; Soares, P.; Ricardo, S.; Cameselle-Teijeiro, J.; Sobrinho-Simões, M. p63 expression in solid cell nests of the thyroid: Further evidence for a stem cell origin. Mod. Pathol. 2003, 16, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Preto, A.; Cameselle-Teijeiro, J.; Moldes-Boullosa, J.; Soares, P.; Cameselle-Teijeiro, J.F.; Silva, P.; Reis-Filho, J.S.; Reyes-Santías, R.M.; Alfonsín-Barreiro, N.; Forteza, J.; et al. Telomerase expression and proliferative activity suggest a stem cell role for thyroid solid cell nests. Mod. Pathol. 2004, 17, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Friedman, S.; Lin, R.Y. Thyroid stem cells: Lessons from normal development and thyroid cancer. Endocr. Relat. Cancer 2008, 15, 51–58. [Google Scholar] [CrossRef]
- Zane, M.; Catalano, V.; Scavo, E.; Bonanno, M.; Pelizzo, M.R.; Todaro, M.; Stassi, G. Estrogens and stem cells in thyroid cancer. Front. Endocrinol. (Lausanne) 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Cui, D.; Xu, S.; Brabant, G.; Derwahl, M. Doxorubicin fails to eradicate cancer stem cells derived from anaplastic thyroid carcinoma cells: Characterization of resistant cells. Int. J. Oncol. 2010, 37, 307–315. [Google Scholar] [PubMed]
- Antico-Arciuch, V.G.; Dima, M.; Liao, X.H.; Refetoff, S.; Di Cristofano, A. Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene 2010, 29, 5678–5686. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chen, G.; Peng, W.; Renko, K.; Derwahl, M. Oestrogen action on thyroid progenitor cells: Relevant for the pathogenesis of thyroid nodules? J. Endocrinol. 2013, 218, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Kouzmenko, A.P.; Takeyama, K.; Ito, S.; Furutani, T.; Sawatsubashi, S.; Maki, A.; Suzuki, E.; Kawasaki, Y.; Akiyama, T.; Tabata, T.; et al. Wnt/β-catenin and estrogen signaling converge in vivo. J. Biol. Chem. 2004, 279, 40255–40258. [Google Scholar] [CrossRef] [PubMed]
- Rajoria, S.; Suriano, R.; Shanmugam, A.; Wilson, Y.L.; Schantz, S.P.; Geliebter, J.; Tiwari, R.K. Metastatic phenotype is regulated by estrogen in thyroid cells. Thyroid 2010, 20, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J.H. Lithium and thyroid. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Urabe, M.; Hershman, J.M.; Pang, X.P.; Murakami, S.; Sugawara, M. Effect of lithium on function and growth of thyroid cells in vitro. Endocrinology 1991, 129, 807–814. [Google Scholar] [CrossRef]
- Rao, A.S.; Kremenevskaja, N.; Resch, J.; Brabant, G. Lithium stimulates proliferation in cultured thyrocytes by activating Wnt/β-catenin signalling. Eur. J. Endocrinol. 2005, 153, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J.H. The effects of lithium therapy on thyroid and thyrotropin-releasing hormone. Thyroid 1998, 8, 909–913. [Google Scholar] [CrossRef]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Blanco, J.; Pednekar, L.; Penas, C.; Li, B.; Martin, V.; Long, J.; Lee, E.; Weiss, W.A.; Rodriguez, C.; Mehrdad, N.; et al. Inhibition of WNT signaling attenuates self-renewal of SHH-subgroup medulloblastoma. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Deiodinase Expression | Normal Function | Hyperthyroidism | Hypothyroidism |
|---|---|---|---|---|
| Thyroid Liver Kidney Skeletal muscle | D1 | Production of T3 into plasma | Increased function | Decreased function |
| Brain | D2 | Production of T3 into plasma and local tissues | Decreased function | Increased function |
| Placenta Brain | D3 | Degradation of T3 | Increased function | Decreased Function |
| Thyroid Cancer | Mutation/Alteration | Frequency | Signaling Pathways | References |
|---|---|---|---|---|
| Papillary Thyroid Carcinoma | BRAF | +++ | MAPK | Nikiforov 2008 [81] Ishigaki 2002 [82] Garcia-Rostan 2001 [74] Rezk 2004 [83] |
| RET/PTC | ++ | PI3K/AKT | ||
| RAS | ++ | MAPK | ||
| PI3K/AKT | ||||
| TRK | + | MAPK | ||
| PI3K/AKT | ||||
| CTNNB1 dysregulation | ++ | Wnt Signaling | ||
| Follicular Thyroid Carcinoma | RAS | +++ | MAPK | Nikiforov 2008 [81] Farrow 2003 [84] Garcia-Rostan 2001 [74] Rezk 2004 [83] |
| PI3K/AKT | ||||
| PAX8-PPARγ | ++ | PI3K/AKT | ||
| PIK3CA | + | PI3K/AKT | ||
| PTEN | + | PI3K/AKT | ||
| CTNNB1 dysregulation | ++ | Wnt Signaling | ||
| Poorly Differentiated Thyroid Carcinoma | RAS | ++ | MAPK | Nikiforov 2008 [81] Garcia-Rostan 2001 [74] |
| PI3K/AKT | ||||
| CTNNB1 | ++ | Wnt signaling | ||
| TP53 | ++ | p53 signaling | ||
| BRAF | ++ | MAPK | ||
| Anaplastic Thyroid Carcinoma | TP53 | +++ | p53 signaling | Nikiforov 2008 [81] Kurihara 2004 [75] Garcia-Rostan 2001 [74] |
| CTNNB1 | +++ | Wnt signaling | ||
| AXIN | +++ | Wnt signaling | ||
| RAS | +++ | MAPK | ||
| PI3K/AKT | ||||
| BRAF | ++ | MAPK |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ely, K.A.; Bischoff, L.A.; Weiss, V.L. Wnt Signaling in Thyroid Homeostasis and Carcinogenesis. Genes 2018, 9, 204. https://doi.org/10.3390/genes9040204
Ely KA, Bischoff LA, Weiss VL. Wnt Signaling in Thyroid Homeostasis and Carcinogenesis. Genes. 2018; 9(4):204. https://doi.org/10.3390/genes9040204
Chicago/Turabian StyleEly, Kim A., Lindsay A. Bischoff, and Vivian L. Weiss. 2018. "Wnt Signaling in Thyroid Homeostasis and Carcinogenesis" Genes 9, no. 4: 204. https://doi.org/10.3390/genes9040204
APA StyleEly, K. A., Bischoff, L. A., & Weiss, V. L. (2018). Wnt Signaling in Thyroid Homeostasis and Carcinogenesis. Genes, 9(4), 204. https://doi.org/10.3390/genes9040204